También podría gustarte
- Vivir con Epilepsia Guía Práctica para Pacientes y FamiliaresDe EverandVivir con Epilepsia Guía Práctica para Pacientes y FamiliaresAún no hay calificaciones
- Comprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerDe EverandComprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerCalificación: 5 de 5 estrellas5/5 (1)
- Enfermedad Motoneurona Inferior y SuperiorDocumento7 páginasEnfermedad Motoneurona Inferior y Superiordianita507Aún no hay calificaciones
- Enfermedad Motoneurona Inferior y Superior - CompressedDocumento7 páginasEnfermedad Motoneurona Inferior y Superior - CompressedLes RAún no hay calificaciones
- Enfermedad Motoneurona Inferior y Superior PDFDocumento7 páginasEnfermedad Motoneurona Inferior y Superior PDFxoxo19881202Aún no hay calificaciones
- Enfermedades NeuromuscularesDocumento23 páginasEnfermedades NeuromuscularesDiego Cruces Bustamante100% (1)
- Lesion Medular No TraumaticaDocumento21 páginasLesion Medular No TraumaticaLuchitoGoicocheaTrauco100% (1)
- Lesión Medular No TraumáticaDocumento21 páginasLesión Medular No TraumáticaRubí BustamanteAún no hay calificaciones
- EF4 Enfermedades DesmielinizantesDocumento14 páginasEF4 Enfermedades DesmielinizantesAdrian GuzmanAún no hay calificaciones
- TEMA 6. Enf. DesmielinizantesDocumento5 páginasTEMA 6. Enf. Desmielinizanteslugmary avellanedaAún no hay calificaciones
- Patología de Motoneurona Superior e Inferior-RehabilitacionDocumento17 páginasPatología de Motoneurona Superior e Inferior-RehabilitacionUlises MontoyaAún no hay calificaciones
- EF4 Enfermedades Desmielinizantes PDFDocumento14 páginasEF4 Enfermedades Desmielinizantes PDFZuleima GómezAún no hay calificaciones
- Esclerosis MultipleDocumento46 páginasEsclerosis MultiplePilar LorenteAún no hay calificaciones
- 28-11 Enf MotoneuronasDocumento6 páginas28-11 Enf Motoneuronasapi-3740897100% (4)
- Enfermedades de Sustancia Blanca - 2Documento16 páginasEnfermedades de Sustancia Blanca - 2Jose Gabriel HerreraAún no hay calificaciones
- Apuntes EnfermedadesDocumento15 páginasApuntes Enfermedadesfrancisca rojas rodriguezAún no hay calificaciones
- ELA ColorDocumento33 páginasELA ColorstylezgzAún no hay calificaciones
- Comisión-Hipercinesias y Trastornos Del Movimiento (Sandra)Documento11 páginasComisión-Hipercinesias y Trastornos Del Movimiento (Sandra)api-3740897100% (1)
- Clase 24 - Patologías SNDocumento38 páginasClase 24 - Patologías SNAlberto Jair Vásquez GonzálezAún no hay calificaciones
- Tomo 5 29-33.en - EsDocumento138 páginasTomo 5 29-33.en - EsMarcela HenaoAún no hay calificaciones
- Esclerosis MúltipleDocumento6 páginasEsclerosis MúltipleFrancis Vásquez PobleteAún no hay calificaciones
- EpilepsiaDocumento39 páginasEpilepsiaSebástian BenitezAún no hay calificaciones
- Trabajo de Patologia EspecializadaDocumento54 páginasTrabajo de Patologia EspecializadaalfredoAún no hay calificaciones
- Treponema Pallidum Es Una Especie Del Género TreponemaDocumento4 páginasTreponema Pallidum Es Una Especie Del Género TreponemajoseAún no hay calificaciones
- Esclerosis MúltipleunqDocumento7 páginasEsclerosis MúltipleunqdanielaAún no hay calificaciones
- Mielina Enfermedad deDocumento6 páginasMielina Enfermedad deAgatha MillerAún no hay calificaciones
- Clase Esclerosis MultipleDocumento15 páginasClase Esclerosis Multipleevelyn millanguirAún no hay calificaciones
- Esclerosis Lateral Amiotrófica: 3º FisioterapiaDocumento13 páginasEsclerosis Lateral Amiotrófica: 3º FisioterapiaAlejandro SuarezAún no hay calificaciones
- Tumores CerebralesDocumento15 páginasTumores CerebralesIsabela RiosAún no hay calificaciones
- Tema 2. Sindrome NefroticoDocumento6 páginasTema 2. Sindrome NefroticoElycardAún no hay calificaciones
- Monografia de Ela Semi TerminadoDocumento25 páginasMonografia de Ela Semi TerminadoHenryGarayarCanchoaAún no hay calificaciones
- ACTIVIDAD 6estudio de Caso Insomnio Familiar FatalDocumento16 páginasACTIVIDAD 6estudio de Caso Insomnio Familiar FatalWILLIAMAún no hay calificaciones
- Esclerosis Lateral AmiotroficaDocumento8 páginasEsclerosis Lateral AmiotroficaNinoskaAlvarezGuzmanAún no hay calificaciones
- EsclerosisDocumento29 páginasEsclerosisjohnAún no hay calificaciones
- Esclerosis MultipleDocumento6 páginasEsclerosis MultipleXiorimar AlbarranAún no hay calificaciones
- Caso ClinicoDocumento7 páginasCaso ClinicoMaritza A. Cunalata OrellanaAún no hay calificaciones
- 4 Bases Biol+ GicasDocumento20 páginas4 Bases Biol+ GicasMaricruz BracamonteAún no hay calificaciones
- El Síndrome de Guillain BarréDocumento8 páginasEl Síndrome de Guillain BarréLeonel VargasAún no hay calificaciones
- Tema 3 Enermedad Neurodegenerativa. Esclerosis Múltiple. Parkinson. Alzheimer. Ela. Corea de Hunghinton, Enfermedad de Creutzfedt-Jakob.Documento43 páginasTema 3 Enermedad Neurodegenerativa. Esclerosis Múltiple. Parkinson. Alzheimer. Ela. Corea de Hunghinton, Enfermedad de Creutzfedt-Jakob.Justin CarpenterAún no hay calificaciones
- LupusDocumento47 páginasLupusLuana FloresAún no hay calificaciones
- Esclerósis Lateral AmiotróficaDocumento21 páginasEsclerósis Lateral AmiotróficaFran GuerreroAún no hay calificaciones
- Mielitis TransversaDocumento7 páginasMielitis Transversa__>killerAún no hay calificaciones
- Esclerosis MultipleDocumento17 páginasEsclerosis MultiplenanandrealinaresAún no hay calificaciones
- Diferencias Entre ELA y EMDocumento3 páginasDiferencias Entre ELA y EMMayte PerejonAún no hay calificaciones
- Final de Neuro 2oo9-2010-1Documento9 páginasFinal de Neuro 2oo9-2010-1tanyilr03Aún no hay calificaciones
- Transtornos NeuromotoresDocumento33 páginasTranstornos Neuromotorescesarabarzua93Aún no hay calificaciones
- Modulo 7 Clase 3 Enfermedades NeurodegenerativasDocumento3 páginasModulo 7 Clase 3 Enfermedades NeurodegenerativasDeyanira S. PicaresAún no hay calificaciones
- Epilepsia PDFDocumento30 páginasEpilepsia PDFluis roseroAún no hay calificaciones
- Que Es La Esclerosis MultipleDocumento13 páginasQue Es La Esclerosis MultipleJesica GiacosaAún no hay calificaciones
- Enfermedades Desmielinizantes PDFDocumento9 páginasEnfermedades Desmielinizantes PDFMarcos TeliasAún no hay calificaciones
- Dialnet EsclerosisMultiple 4018455 PDFDocumento20 páginasDialnet EsclerosisMultiple 4018455 PDFholaAún no hay calificaciones
- Enfermedades de La Neurona MotoraDocumento27 páginasEnfermedades de La Neurona MotoraStefany DiazAún no hay calificaciones
- 8 - Esclerosis Lateral AmiotróficaDocumento11 páginas8 - Esclerosis Lateral AmiotróficaErmilo José Echeverría OrtegónAún no hay calificaciones
- Parcial Neurodesarrollo 2do CuatrimestreDocumento14 páginasParcial Neurodesarrollo 2do CuatrimestreLuana RoblesAún no hay calificaciones
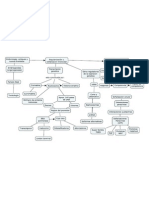
- Mapa NeuroDocumento1 páginaMapa NeuroMontse VázquezAún no hay calificaciones
- 2013 Ela emDocumento38 páginas2013 Ela emjuanoscopioAún no hay calificaciones
- Sindrome de Guillain Barre ResumenDocumento8 páginasSindrome de Guillain Barre ResumenAlisol100% (1)
- Propiedades de los derivados del cannabis en el AlzheimerDe EverandPropiedades de los derivados del cannabis en el AlzheimerAún no hay calificaciones
- Genereralidades Del Tejido MuscularDocumento28 páginasGenereralidades Del Tejido MuscularSamuel GasparAún no hay calificaciones
- Hiper e HiporreflexiaDocumento4 páginasHiper e HiporreflexiaSamuel GasparAún no hay calificaciones
- Sindrome MotoneuronaDocumento5 páginasSindrome MotoneuronaSamuel GasparAún no hay calificaciones
- Ivu (1) 2Documento36 páginasIvu (1) 2Samuel GasparAún no hay calificaciones
- Actualizaciones AMIR 2020 Covid-19Documento9 páginasActualizaciones AMIR 2020 Covid-19Dina Eunise100% (1)
- TORCHDocumento2 páginasTORCHSamuel GasparAún no hay calificaciones
- Emergencias-2002 14 1 S19-27 PDFDocumento9 páginasEmergencias-2002 14 1 S19-27 PDFjjjkkAún no hay calificaciones
- Receta Nieve de GarrafaDocumento1 páginaReceta Nieve de GarrafaGerardo Valdez50% (2)
- Articulo PulmonarDocumento26 páginasArticulo PulmonarSamuel GasparAún no hay calificaciones
- Primer Capitulo Embriologia - EmbrioDocumento1 páginaPrimer Capitulo Embriologia - EmbrioSamuel GasparAún no hay calificaciones
- Riñón 3Documento26 páginasRiñón 3Rodrigo S. AvilésAún no hay calificaciones
- Clase 6 - Shiatsu - CiriaxDocumento22 páginasClase 6 - Shiatsu - CiriaxJohanLiebertAún no hay calificaciones
- Intercambio Con Un Ingles PDFDocumento24 páginasIntercambio Con Un Ingles PDFEugenia Quintero0% (2)
- Trabajo Entregable 1era EntregaDocumento22 páginasTrabajo Entregable 1era EntregaEder Edu Silva SilvaAún no hay calificaciones
- 830curso Gaviota tcm7-525561Documento12 páginas830curso Gaviota tcm7-525561NORCABRERAAún no hay calificaciones
- Elaboracion de Proyectos de InversionDocumento31 páginasElaboracion de Proyectos de InversionJuliana Aguirre S.100% (1)
- Ejemplo de IndiceDocumento10 páginasEjemplo de IndiceRafael QuichuAún no hay calificaciones
- Tarea 1 Geografia Dominicana IDocumento6 páginasTarea 1 Geografia Dominicana ICarlos Encarnacion VicenteAún no hay calificaciones
- Power ScopeDocumento16 páginasPower ScopeMarianela Ruiz TeresaAún no hay calificaciones
- Hoja de Calculo de ConsolidacionDocumento50 páginasHoja de Calculo de ConsolidacionjuanAún no hay calificaciones
- Q1co Hse Ir Sacyr-A PD 23.04.22Documento9 páginasQ1co Hse Ir Sacyr-A PD 23.04.22juan carlos huallpa espinozaAún no hay calificaciones
- ENyD Inteligencia Artificial - F. DoralDocumento22 páginasENyD Inteligencia Artificial - F. DoralMonica AriasAún no hay calificaciones
- Techo Básico para Familias VulnerablesDocumento13 páginasTecho Básico para Familias Vulnerablesroberto alexander monterrosa figueroaAún no hay calificaciones
- EngranajesDocumento11 páginasEngranajesEdmundo GonzalezAún no hay calificaciones
- Las Desigualdades o Inecuaciones y Sus PropiedadesDocumento38 páginasLas Desigualdades o Inecuaciones y Sus PropiedadesBrayeanSilvaSanchezAún no hay calificaciones
- Adecuaciones y Proteccion ArticularDocumento50 páginasAdecuaciones y Proteccion ArticularAle PavezAún no hay calificaciones
- PACO DE LUCIA-al-yazirat2014Documento52 páginasPACO DE LUCIA-al-yazirat2014Diego Morilla100% (1)
- Pauta Certamen 2Documento5 páginasPauta Certamen 2Daniel Antonio Jelvez CifuentesAún no hay calificaciones
- Plan ContingenciasDocumento80 páginasPlan ContingenciasWILLIAM JOHNY JAVIER ARENASAún no hay calificaciones
- WORD1Documento5 páginasWORD1Jhonatan choqueAún no hay calificaciones
- Tarea 2Documento7 páginasTarea 2brayanbAún no hay calificaciones
- Liahona Mayo 2012Documento148 páginasLiahona Mayo 2012intrabytAún no hay calificaciones
- Ensayo. Trabajo y Enajenación en La Sociedad ActualDocumento12 páginasEnsayo. Trabajo y Enajenación en La Sociedad ActualMaría LuqueAún no hay calificaciones
- 3 - Derivadas Compuestas e ImplicitasDocumento25 páginas3 - Derivadas Compuestas e ImplicitasAgustin VargasAún no hay calificaciones
- Investigacion de CampoDocumento7 páginasInvestigacion de CampoJose MoraAún no hay calificaciones
- 4 Tinigua - Corredor Andino Orinocense-AmazonicoDocumento9 páginas4 Tinigua - Corredor Andino Orinocense-AmazonicocarlosAún no hay calificaciones
- Máquinas de Soporte Vectorial (SVM)Documento12 páginasMáquinas de Soporte Vectorial (SVM)Luis Enrique Méndez Costales100% (1)
- Onfalocele - GastrosquisisDocumento40 páginasOnfalocele - GastrosquisisLuis Arias86% (7)
- Kaleidoscopio 30 Version DigitalDocumento61 páginasKaleidoscopio 30 Version DigitalLuis Alberto Castañeda RojasAún no hay calificaciones
- Polarizacion Del Transistor BJTDocumento29 páginasPolarizacion Del Transistor BJTLarry Christian Tumialán BorjaAún no hay calificaciones