También podría gustarte
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleAún no hay calificaciones
- Anatomía, fisiología y fisiopatología renalDocumento21 páginasAnatomía, fisiología y fisiopatología renalMax Edwin Gorena EspinozaAún no hay calificaciones
- Insuficiencia Renal Cronica+2022Documento43 páginasInsuficiencia Renal Cronica+2022Vanessa AgurtoAún no hay calificaciones
- Clase 3Documento20 páginasClase 3MARIA JOSEFINA GUILLARD HERRERAAún no hay calificaciones
- ERC Interna 2Documento8 páginasERC Interna 2Ainhoa UrlézagaAún no hay calificaciones
- Patología del Sistema UrinarioDocumento52 páginasPatología del Sistema UrinarioDeny Juliana ZAPATA CARDONA0% (1)
- Sindrome Nefrotico, Nefritico, Falla RenalDocumento32 páginasSindrome Nefrotico, Nefritico, Falla RenalweplexAún no hay calificaciones
- Insuficiencia Renal Crónica (Irc) : Dr. Manuel Silva Zumarán Uap - HNDMDocumento25 páginasInsuficiencia Renal Crónica (Irc) : Dr. Manuel Silva Zumarán Uap - HNDMOCTAVIOAún no hay calificaciones
- I.R.A y C-3Documento54 páginasI.R.A y C-3Fiore RamosAún no hay calificaciones
- Insuficiencia Renal AgudaDocumento9 páginasInsuficiencia Renal AgudaAlexandra PiñeroAún no hay calificaciones
- 2.falla Renal - Diálisis Y Terapia de Reemplazo RenalDocumento7 páginas2.falla Renal - Diálisis Y Terapia de Reemplazo RenalJOSEFA MARCELA SANHUEZA GONZALEZAún no hay calificaciones
- UABP 6 - Insuficiencia Renal CronicaDocumento4 páginasUABP 6 - Insuficiencia Renal CronicaRosa MarinelliAún no hay calificaciones
- Insuficiencia Renal AgudaDocumento57 páginasInsuficiencia Renal AgudaAle Saucedo HernandezAún no hay calificaciones
- Sindromes RenalesDocumento4 páginasSindromes RenalesElia SalazarAún no hay calificaciones
- IRC: Insuficiencia Renal CrónicaDocumento56 páginasIRC: Insuficiencia Renal CrónicaAmerc CuscoAún no hay calificaciones
- Resumos RenalDocumento113 páginasResumos RenalalvaroAún no hay calificaciones
- Pato 7.1 - RenalDocumento6 páginasPato 7.1 - RenaldanielamcgroffAún no hay calificaciones
- Insuficiencia Renal Crónica: Factores de Riesgo y Manifestaciones (IRCDocumento34 páginasInsuficiencia Renal Crónica: Factores de Riesgo y Manifestaciones (IRCPaloma JGAún no hay calificaciones
- Insuficiencia RenalDocumento89 páginasInsuficiencia Renalvladi pizarroAún no hay calificaciones
- Cuidados del paciente en hemodiálisis: generalidades de terapia dialíticaDocumento35 páginasCuidados del paciente en hemodiálisis: generalidades de terapia dialíticaJosefa MaureiraAún no hay calificaciones
- Patología del sistema urinario: Riñón y tracto urinarioDocumento43 páginasPatología del sistema urinario: Riñón y tracto urinarioVictorGimenesRiveroAún no hay calificaciones
- ENFERMEDAD RENAL CRÓNICA scribdDocumento10 páginasENFERMEDAD RENAL CRÓNICA scribdAriana AguirreAún no hay calificaciones
- HistologiaDocumento14 páginasHistologiaGime GimeAún no hay calificaciones
- Urologia .... - 1 ExposiciónDocumento60 páginasUrologia .... - 1 Exposicióncarlos valderramaAún no hay calificaciones
- Clase 4. Alteraciones TubularesDocumento4 páginasClase 4. Alteraciones TubularesJavier RodriguezAún no hay calificaciones
- 2 Clase Sindromes ClinicosDocumento90 páginas2 Clase Sindromes ClinicosVitrena StoreAún no hay calificaciones
- Etiologia y Fisiopatologia: Se Dividen Las Causas en Tres Categorías: HiperazoemiaDocumento6 páginasEtiologia y Fisiopatologia: Se Dividen Las Causas en Tres Categorías: HiperazoemiaCATALINA IGNACIA ASPEE CARRASCOAún no hay calificaciones
- Pre Internado 2021 - Villamemo NefrologíaDocumento10 páginasPre Internado 2021 - Villamemo NefrologíaANTOLINO GENARO ROSALES PIJOAún no hay calificaciones
- Insuficiencia RenalDocumento5 páginasInsuficiencia RenalToni TeseoAún no hay calificaciones
- Renal BolilleroDocumento17 páginasRenal BolilleroNahuel ColmanAún no hay calificaciones
- Insuficiencia RenalDocumento30 páginasInsuficiencia Renaljenny machacaAún no hay calificaciones
- P.Nefro INSUFICIENCIA RENAL 3Documento5 páginasP.Nefro INSUFICIENCIA RENAL 3Valeria Jadue MorónAún no hay calificaciones
- ERC JorgeDocumento50 páginasERC JorgeJorge Vasquez ZambranoAún no hay calificaciones
- RenalDocumento11 páginasRenaljanny RomeroAún no hay calificaciones
- Insuficiencia Renal CrónicaDocumento4 páginasInsuficiencia Renal CrónicaJunior A. SanchezAún no hay calificaciones
- IRA: causas, diagnóstico y tratamientoDocumento8 páginasIRA: causas, diagnóstico y tratamientoAle OsorioAún no hay calificaciones
- Falla Renal CrónicaDocumento15 páginasFalla Renal CrónicaFernanda RodriguezAún no hay calificaciones
- Insuficiencia Renal AgudaDocumento31 páginasInsuficiencia Renal AgudaLenny NogueraAún no hay calificaciones
- IRC: insuficiencia renal crónicaDocumento10 páginasIRC: insuficiencia renal crónicaBellerlynAún no hay calificaciones
- Nefropatías Túbulo IntersticialesDocumento13 páginasNefropatías Túbulo IntersticialesPoPe MendeleroAún no hay calificaciones
- 1 'Preguntas Clinicas de Peq'Documento42 páginas1 'Preguntas Clinicas de Peq'Gustavo BritoAún no hay calificaciones
- 1insuficiencia Renal AgudaDocumento20 páginas1insuficiencia Renal AgudaDaniel HernandezAún no hay calificaciones
- Insuficiencia Renal Crónica: Causas, Etapas y Alteraciones MetabólicasDocumento23 páginasInsuficiencia Renal Crónica: Causas, Etapas y Alteraciones MetabólicasMilan Montilla100% (1)
- 04 Irc-1Documento54 páginas04 Irc-1Marcos Anderson Gomez CaleroAún no hay calificaciones
- Fisiopatología y Patogenia General Del Rión y de Las Vías UrinariasDocumento13 páginasFisiopatología y Patogenia General Del Rión y de Las Vías UrinariasVil Lanego TiAún no hay calificaciones
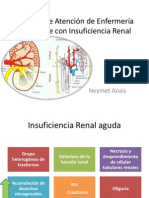
- Proceso de atención en paciente con insuficiencia renalDocumento43 páginasProceso de atención en paciente con insuficiencia renalpiper0blueAún no hay calificaciones
- Irc IraDocumento3 páginasIrc IraAriel SiguenzaAún no hay calificaciones
- Enfermedad Renal AgudaDocumento38 páginasEnfermedad Renal AgudaLiliana Altamirano SoteloAún no hay calificaciones
- Teorico RenalDocumento43 páginasTeorico RenalMabel TorrezAún no hay calificaciones
- TAREA 2 PropeDocumento6 páginasTAREA 2 PropedaelAún no hay calificaciones
- EDEMA Y EFUSIÓN: CAUSAS, TIPOS E INSUFICIENCIA RENALDocumento84 páginasEDEMA Y EFUSIÓN: CAUSAS, TIPOS E INSUFICIENCIA RENALRuben Apaza ApazaAún no hay calificaciones
- Clase 03 Pruebas - de - Funcion - Renal - y - PatologiasDocumento50 páginasClase 03 Pruebas - de - Funcion - Renal - y - PatologiasRENZO ARCHIE ALBERTI CHOMONAún no hay calificaciones
- Sindrome UremicoDocumento54 páginasSindrome UremicoHumberto CastroAún no hay calificaciones
- Hiperparatiroidismo: causas, manifestaciones y tratamientoDocumento6 páginasHiperparatiroidismo: causas, manifestaciones y tratamientoliliana delgadoAún no hay calificaciones
- Conceptos Medicos y EnfermedadesDocumento11 páginasConceptos Medicos y EnfermedadesPaula PerezAún no hay calificaciones
- Lesion Renal AgudaDocumento25 páginasLesion Renal Agudaangie chamorroAún no hay calificaciones
- Insuficiencia Renal CronicaDocumento146 páginasInsuficiencia Renal CronicaLupita Padilla SánchezAún no hay calificaciones
- Renal Fisiopatología UnabDocumento22 páginasRenal Fisiopatología UnabPetter ZapatoAún no hay calificaciones
- Fisiopatología Renal FCV UCVDocumento33 páginasFisiopatología Renal FCV UCVAkane UchihaAún no hay calificaciones
- Insuficiencia Renal Aguda (8.2)Documento5 páginasInsuficiencia Renal Aguda (8.2)Eduardo FreireAún no hay calificaciones
- Clase 8Documento5 páginasClase 8Valen ViolaAún no hay calificaciones
- Clase 9Documento4 páginasClase 9Valen ViolaAún no hay calificaciones
- Estadística médica: Chi cuadrado y riesgosDocumento3 páginasEstadística médica: Chi cuadrado y riesgosValen ViolaAún no hay calificaciones
- Clase 7Documento3 páginasClase 7Valen ViolaAún no hay calificaciones
- Clase 6Documento5 páginasClase 6Valen ViolaAún no hay calificaciones
- Clase 3Documento4 páginasClase 3Valen ViolaAún no hay calificaciones
- Clase 4Documento3 páginasClase 4Valen ViolaAún no hay calificaciones
- Clase 11Documento2 páginasClase 11Valen ViolaAún no hay calificaciones
- Clase 2Documento5 páginasClase 2Valen ViolaAún no hay calificaciones
- Estadística médica: Inferencia estadísticaDocumento4 páginasEstadística médica: Inferencia estadísticaValen ViolaAún no hay calificaciones
- Clase 1Documento7 páginasClase 1Valen ViolaAún no hay calificaciones
- Patologia OseaDocumento10 páginasPatologia OseaValen ViolaAún no hay calificaciones
- Cardiologia 11 MIR PDFDocumento172 páginasCardiologia 11 MIR PDFCarlos Paulino TaverasAún no hay calificaciones
- Enteroparasitosis PDFDocumento54 páginasEnteroparasitosis PDFValen ViolaAún no hay calificaciones
- Triptico SuicidioDocumento2 páginasTriptico SuicidioSaira Hernandez MoraAún no hay calificaciones
- Historia ClínicaDocumento3 páginasHistoria ClínicaCentro de Salud Villa Mariano Matamoros IMSSAún no hay calificaciones
- 1-Complicaciones Graves de La HASDocumento39 páginas1-Complicaciones Graves de La HASGerardo Alberto Fernández LunaAún no hay calificaciones
- Epiteliais: FundamentalesDocumento43 páginasEpiteliais: FundamentalesayllaAún no hay calificaciones
- Anemia de La Enfermedad CronicaDocumento20 páginasAnemia de La Enfermedad CronicaJUAN DIEGO TUCTO HERNÁNDEZAún no hay calificaciones
- Clase Nro 7Documento23 páginasClase Nro 7Terapia Fisica Mejora JtlAún no hay calificaciones
- Guion de Un Podcast Sobre La Vida SaludableDocumento3 páginasGuion de Un Podcast Sobre La Vida SaludableJoshua Rivas Llanos0% (1)
- EsclerodermiaDocumento15 páginasEsclerodermiaRita CássiaAún no hay calificaciones
- Adobe Scan 13 Nov. 2021Documento1 páginaAdobe Scan 13 Nov. 2021Marcelino BaenaAún no hay calificaciones
- Ups Nna-EstrategiasDocumento5 páginasUps Nna-EstrategiasJohanna PinillosAún no hay calificaciones
- Papilomatosis Confluente y ReticuladaDocumento3 páginasPapilomatosis Confluente y ReticuladaJuan Manuel Morón OcañaAún no hay calificaciones
- DiabetesDocumento6 páginasDiabetesLiz Marie FarrugiaAún no hay calificaciones
- Crecimiento y Desarrollo Del Prescolar y EscolarDocumento35 páginasCrecimiento y Desarrollo Del Prescolar y EscolarMariaAún no hay calificaciones
- Tarea 4 Semiologia ColaborativoDocumento10 páginasTarea 4 Semiologia Colaborativoerika vanessa macias irreñoAún no hay calificaciones
- Caja Roja y Rosa 421904 Downloable 2995420Documento6 páginasCaja Roja y Rosa 421904 Downloable 2995420MARGOTH YADIRA ESCAMILLA TORRESAún no hay calificaciones
- Evaluación nutricional y valoración geriátrica integral en adultos mayoresDocumento46 páginasEvaluación nutricional y valoración geriátrica integral en adultos mayoresMARIEL ADALI HERRERA MARCHENAAún no hay calificaciones
- Album FarmacologicoDocumento26 páginasAlbum FarmacologicokaryAún no hay calificaciones
- La Pesquisa Neonatal o Prueba Del TalónDocumento3 páginasLa Pesquisa Neonatal o Prueba Del TalónCris GalantónAún no hay calificaciones
- Hiv Pediatrico Algoritmo Diagnostico NotificacionDocumento1 páginaHiv Pediatrico Algoritmo Diagnostico NotificacionJuanMa ParadaAún no hay calificaciones
- EJSSP2A2Documento4 páginasEJSSP2A2Eduardo Jacobo Santos SalasAún no hay calificaciones
- Catalogo Biomedical 1Documento135 páginasCatalogo Biomedical 1ISAIAS ISAAC SALAS CARHUAMACAAún no hay calificaciones
- Examen 1 Resuelto MicroDocumento8 páginasExamen 1 Resuelto MicroAdriana Cisneros MontalvoAún no hay calificaciones
- Caso Clínico N°4Documento40 páginasCaso Clínico N°4Tatiana AquinoAún no hay calificaciones
- HEPATITISDocumento9 páginasHEPATITISalexandra palmaAún no hay calificaciones
- Caso Clinico Insuficiencia Renal AgudaDocumento8 páginasCaso Clinico Insuficiencia Renal AgudaDenisse FloresAún no hay calificaciones
- Epidemiología GlosarioDocumento3 páginasEpidemiología GlosarioMariaEsther MuñozGomezAún no hay calificaciones
- Neuro Progreso 1Documento127 páginasNeuro Progreso 1Joseph AndradeAún no hay calificaciones
- Documento de Consenso Sobre Abordaje D Ela Enfermedad de Chagas en Atencion Primaria de Salud de Areas No Endemicas Roca 2015Documento10 páginasDocumento de Consenso Sobre Abordaje D Ela Enfermedad de Chagas en Atencion Primaria de Salud de Areas No Endemicas Roca 2015Adriana Santander OliveraAún no hay calificaciones
- Anexo Plazos de Espera - Póliza IndividualDocumento2 páginasAnexo Plazos de Espera - Póliza IndividualJulio Camejo CarrilloAún no hay calificaciones
- RCP PediatricaDocumento42 páginasRCP PediatricajamesAún no hay calificaciones
- El libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)De EverandEl libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)Calificación: 3 de 5 estrellas3/5 (2)
- Terapia cognitiva: Conceptos básicos y profundizaciónDe EverandTerapia cognitiva: Conceptos básicos y profundizaciónCalificación: 5 de 5 estrellas5/5 (1)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- TDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosDe EverandTDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosCalificación: 4 de 5 estrellas4/5 (8)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)
- Neuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaDe EverandNeuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaCalificación: 4 de 5 estrellas4/5 (16)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- Fisiopatología de las enfermedades cardiovascularesDe EverandFisiopatología de las enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)
- Dieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasDe EverandDieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasCalificación: 4 de 5 estrellas4/5 (9)
- Gua Sha: Guía de autotratamiento completoDe EverandGua Sha: Guía de autotratamiento completoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- La Dieta Mediterránea Para Principiantes, Guía Paso A Paso Con Recetas Para Comer Mejor Y Bajar De PesoDe EverandLa Dieta Mediterránea Para Principiantes, Guía Paso A Paso Con Recetas Para Comer Mejor Y Bajar De PesoCalificación: 5 de 5 estrellas5/5 (2)
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- Prescripción de ejercico físico para la saludDe EverandPrescripción de ejercico físico para la saludCalificación: 5 de 5 estrellas5/5 (1)
- Psicópatas seriales: Un recorrido por su oscura e inquietante naturalezaDe EverandPsicópatas seriales: Un recorrido por su oscura e inquietante naturalezaCalificación: 4 de 5 estrellas4/5 (3)
- Genética general: Libro de textoDe EverandGenética general: Libro de textoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- ABC de los procedimientos médicos básicos: Una guía de aprendizaje y enseñanza para profesionales de medicinaDe EverandABC de los procedimientos médicos básicos: Una guía de aprendizaje y enseñanza para profesionales de medicinaCalificación: 4 de 5 estrellas4/5 (4)
- La cocina ayurvédica: Recetas para la salud y el bienestarDe EverandLa cocina ayurvédica: Recetas para la salud y el bienestarCalificación: 5 de 5 estrellas5/5 (6)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Más Aristóteles y menos Concerta®: Las cuatro causas del TDAHDe EverandMás Aristóteles y menos Concerta®: Las cuatro causas del TDAHCalificación: 5 de 5 estrellas5/5 (2)
- Psicoterapia breve con niños y adolescentesDe EverandPsicoterapia breve con niños y adolescentesCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Psiconeuroinmunología para la práctica clínicaDe EverandPsiconeuroinmunología para la práctica clínicaCalificación: 5 de 5 estrellas5/5 (4)
- La acupuntura para prevenir y curar las enfermedadesDe EverandLa acupuntura para prevenir y curar las enfermedadesAún no hay calificaciones
- Cardiología y enfermedades cardiovascularesDe EverandCardiología y enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)