También podría gustarte
- UF0352 - Acondicionamiento de la carne para su comercializaciónDe EverandUF0352 - Acondicionamiento de la carne para su comercializaciónAún no hay calificaciones
- Acondicionamiento de la carne para su comercialización. INAI0108De EverandAcondicionamiento de la carne para su comercialización. INAI0108Aún no hay calificaciones
- Practica 11 BromatologiaDocumento19 páginasPractica 11 BromatologiaANTUANETHE LETICIA OSORES LAUREANOAún no hay calificaciones
- Tema 5 CARNES Y DERIVADOS1Documento16 páginasTema 5 CARNES Y DERIVADOS1Jakelin MandujanoAún no hay calificaciones
- Analisis Fisico Quimico en CarnesDocumento4 páginasAnalisis Fisico Quimico en CarnesLisbeth Miyoun Sunhg71% (7)
- CarnessssDocumento17 páginasCarnessssJoseinAceroCrispinAún no hay calificaciones
- Informe BIO Lab FinalDocumento10 páginasInforme BIO Lab Finalfernandanavarro.ffAún no hay calificaciones
- Análisis de Calidad en Muestras de CarneDocumento5 páginasAnálisis de Calidad en Muestras de CarneHernan Tamara OrtegaAún no hay calificaciones
- Taller 24 de Marzo-Las Materias Primas - ResueltoDocumento7 páginasTaller 24 de Marzo-Las Materias Primas - ResueltoAndres Juan TorresAún no hay calificaciones
- Analisis Fisico Quimico en CarnesDocumento4 páginasAnalisis Fisico Quimico en CarnesYaneth ChucuyaAún no hay calificaciones
- Curado de CarnesDocumento20 páginasCurado de CarnesJulio César Barbarán DíazAún no hay calificaciones
- Bromatologia Carne OriginalDocumento13 páginasBromatologia Carne Originalestefany villcaAún no hay calificaciones
- Bromatologia Carne AnalisisDocumento13 páginasBromatologia Carne Analisisestefany villcaAún no hay calificaciones
- Resumenes Tecno 3Documento8 páginasResumenes Tecno 3diegoAún no hay calificaciones
- EscaldadoDocumento3 páginasEscaldadodarioAún no hay calificaciones
- Practica N.11 BromatologiaDocumento11 páginasPractica N.11 Bromatologiarogger castro100% (1)
- Práctica 3Documento8 páginasPráctica 3Ruth LCAún no hay calificaciones
- Informe Lab CarneDocumento7 páginasInforme Lab Carneanon_232847364Aún no hay calificaciones
- Componente Práctico Virtual - Angela - RamírezDocumento34 páginasComponente Práctico Virtual - Angela - Ramírezangela ramirezAún no hay calificaciones
- Comportamiento y Determinación de Acidez y PH en Carne de PavitaDocumento6 páginasComportamiento y Determinación de Acidez y PH en Carne de PavitaJairo Hernan RodríguezAún no hay calificaciones
- Analisis EmbutidoDocumento8 páginasAnalisis EmbutidoOLGA VILLADIEGOAún no hay calificaciones
- Capítulo IV - Operaciones Básicas y Complementarias en La Elaboración de EmbutidosDocumento38 páginasCapítulo IV - Operaciones Básicas y Complementarias en La Elaboración de Embutidosjorge macedoAún no hay calificaciones
- Que Son Las Carnes Blancas Trabajo Emanuel)Documento7 páginasQue Son Las Carnes Blancas Trabajo Emanuel)Emanuel Zuleta MolinaAún no hay calificaciones
- Propiedades Funcionales de La CarneDocumento13 páginasPropiedades Funcionales de La CarneLuzSmeli50% (2)
- Identificacion de CarnesDocumento91 páginasIdentificacion de CarnesIsra Hernandez100% (2)
- Operaciones Básicas y Complementarias en La Elaboración de EmbutidosDocumento38 páginasOperaciones Básicas y Complementarias en La Elaboración de EmbutidosJackelineAún no hay calificaciones
- Análisis de La Carne BrailynDocumento6 páginasAnálisis de La Carne BrailynAndrea Paola Diaz BuelvasAún no hay calificaciones
- CarnesDocumento20 páginasCarnesPaola Flores VasquezAún no hay calificaciones
- Carne Definiciones Legales y Maduración de La CarneDocumento32 páginasCarne Definiciones Legales y Maduración de La CarneLizeth LoubetAún no hay calificaciones
- Area de Congelado y OreoDocumento15 páginasArea de Congelado y OreoRocio Milagros Farfan SilvaAún no hay calificaciones
- Asignatura: Ingeniria Y Tecnologia de Productos CarnicosDocumento51 páginasAsignatura: Ingeniria Y Tecnologia de Productos Carnicosjuan alarcon berrocalAún no hay calificaciones
- Organolépticas de Las CarnesDocumento4 páginasOrganolépticas de Las CarnesLaura CamilaAún no hay calificaciones
- PRACTICA #9 CarneDocumento4 páginasPRACTICA #9 CarneElizabeth Pereira FariñaAún no hay calificaciones
- Determinación de Cra y Ce en La Carne FrescaDocumento12 páginasDeterminación de Cra y Ce en La Carne FrescaHOLA BUEN DIA0% (1)
- CarneDocumento45 páginasCarnecacua30100% (1)
- Chorizo Informe Final IMPRIMIRDocumento21 páginasChorizo Informe Final IMPRIMIRJunior Galvez VenegasAún no hay calificaciones
- Análisis de La CarneDocumento6 páginasAnálisis de La CarneMaryori Yadira CMAún no hay calificaciones
- Practica 4 Caracterizacion Fisicoquimica de La Carne FrescaDocumento12 páginasPractica 4 Caracterizacion Fisicoquimica de La Carne FrescaIbeth Anny Coavoy SánchezAún no hay calificaciones
- Maduracion Carne de BovinoDocumento8 páginasMaduracion Carne de BovinoJosé MontalvoAún no hay calificaciones
- Embutidos de TruchaDocumento7 páginasEmbutidos de TruchaEdison Cayo Condori100% (2)
- Proyecto de La Refrigeración de La CarneDocumento5 páginasProyecto de La Refrigeración de La CarneRina Licet Socompi AguilarAún no hay calificaciones
- Catacion de CarneDocumento7 páginasCatacion de CarnegrimaldoAún no hay calificaciones
- Instructivo Practica 6 Industria de La CarneDocumento4 páginasInstructivo Practica 6 Industria de La CarneClau GarcíaAún no hay calificaciones
- Manual Practico de Productos CarnicosDocumento70 páginasManual Practico de Productos CarnicosKristoper G. Mendo0% (1)
- La Carne Se Suele Analizar para Indicar Niveles de Frescura o Determinar Si Está RanciaDocumento2 páginasLa Carne Se Suele Analizar para Indicar Niveles de Frescura o Determinar Si Está RanciaEstuardo CastilloAún no hay calificaciones
- Métodos de Maduración y Conservacion de Las Carnes RojasDocumento2 páginasMétodos de Maduración y Conservacion de Las Carnes RojasSantiago Bermudez SierraAún no hay calificaciones
- Analisis de CarnesDocumento17 páginasAnalisis de CarnesNury Marquez Alvis100% (2)
- BR Carne y Derivados-Pescado y Derivados - HuevosDocumento70 páginasBR Carne y Derivados-Pescado y Derivados - HuevosRocyta Caceres PerezAún no hay calificaciones
- 01.determinacion de Humedad, PH y Acidez en Productos CarnicosDocumento13 páginas01.determinacion de Humedad, PH y Acidez en Productos CarnicosEmmanuel Mendoza100% (1)
- Calidad de La CarneDocumento10 páginasCalidad de La CarneRafael Angel Huiza TrujilloAún no hay calificaciones
- Lab 150109131316 Conversion Gate02Documento38 páginasLab 150109131316 Conversion Gate02Marcos Eduardo Pineda GarridoAún no hay calificaciones
- Cuaderno de Prácticas de Productos Carnicos 21-22Documento20 páginasCuaderno de Prácticas de Productos Carnicos 21-22veronicaAún no hay calificaciones
- Maduración de La Carne, Un Proceso DesconocidoDocumento6 páginasMaduración de La Carne, Un Proceso Desconocidoluis pablo SistiAún no hay calificaciones
- Elaboración de Chorizo CriolloDocumento9 páginasElaboración de Chorizo CriolloRicardo Patiño67% (3)
- Determinacion de La Acides en La CarneDocumento11 páginasDeterminacion de La Acides en La CarneLiz MamaniAún no hay calificaciones
- 4to Informe de Laboratorio de Ciencias de La Carne Trabajo Terminado y EntregadoDocumento12 páginas4to Informe de Laboratorio de Ciencias de La Carne Trabajo Terminado y EntregadoJose Cesar MamaniAún no hay calificaciones
- Hot DogDocumento35 páginasHot DogFatima Flores MunaycoAún no hay calificaciones
- Acondicionamiento de la carne para su uso industrial. INAI0108De EverandAcondicionamiento de la carne para su uso industrial. INAI0108Aún no hay calificaciones
- Lo mejor de la cocina Árabe en tu mesaDe EverandLo mejor de la cocina Árabe en tu mesaCalificación: 5 de 5 estrellas5/5 (1)
- Pollo Gourmet - Consigue El Sabor Gourmet En Tus Comidas Diarias. Descubre El Sabor Gourmet Con Recetas de Pollo Economicas, Saludables Y ExquisitasDe EverandPollo Gourmet - Consigue El Sabor Gourmet En Tus Comidas Diarias. Descubre El Sabor Gourmet Con Recetas de Pollo Economicas, Saludables Y ExquisitasCalificación: 5 de 5 estrellas5/5 (1)
- Glosario de Farmacia HospitalariaDocumento10 páginasGlosario de Farmacia HospitalariaGenesis Fajardo IgotsevenAún no hay calificaciones
- Organigrama de Farmacia HospitalariaDocumento2 páginasOrganigrama de Farmacia HospitalariaGenesis Fajardo Igotseven100% (1)
- Resumen de Ruta de Medicamentos e InsumosDocumento3 páginasResumen de Ruta de Medicamentos e InsumosGenesis Fajardo IgotsevenAún no hay calificaciones
- Ejemplos de Medicamentos BasicosDocumento4 páginasEjemplos de Medicamentos BasicosGenesis Fajardo IgotsevenAún no hay calificaciones
- Tarea Grupal N°1 - Registro Sanitario de Productos Cosméticos en Ecuador - Química CosméticaDocumento16 páginasTarea Grupal N°1 - Registro Sanitario de Productos Cosméticos en Ecuador - Química CosméticaGenesis Fajardo IgotsevenAún no hay calificaciones
- Práctica 2 Química AnalíticaDocumento6 páginasPráctica 2 Química AnalíticaGenesis Fajardo IgotsevenAún no hay calificaciones
- Trabajo de Investigación - Formulación de Una Crema Reafirmante de Cara y CuelloDocumento9 páginasTrabajo de Investigación - Formulación de Una Crema Reafirmante de Cara y CuelloGenesis Fajardo IgotsevenAún no hay calificaciones
- Informe 3 - Analisis de Frutas DeshidratadasDocumento4 páginasInforme 3 - Analisis de Frutas DeshidratadasGenesis Fajardo IgotsevenAún no hay calificaciones
- Práctica 1 Química AnalíticaDocumento8 páginasPráctica 1 Química AnalíticaGenesis Fajardo IgotsevenAún no hay calificaciones
- Buenas Practicas de ManufacturaDocumento3 páginasBuenas Practicas de ManufacturaGenesis Fajardo IgotsevenAún no hay calificaciones
- Práctica 2 Química AnalíticaDocumento7 páginasPráctica 2 Química AnalíticaGenesis Fajardo IgotsevenAún no hay calificaciones
- Importancia de La InocuidadDocumento2 páginasImportancia de La InocuidadGenesis Fajardo IgotsevenAún no hay calificaciones
- Clasificación de Flores y FrutosDocumento8 páginasClasificación de Flores y FrutosGenesis Fajardo IgotsevenAún no hay calificaciones
- Reactivo de BiuretDocumento1 páginaReactivo de BiuretGenesis Fajardo Igotseven100% (1)
- Clostridium BotulinumDocumento3 páginasClostridium BotulinumGenesis Fajardo IgotsevenAún no hay calificaciones
- Clasificacion de Los Metodos AnaliticosDocumento1 páginaClasificacion de Los Metodos AnaliticosGenesis Fajardo Igotseven100% (1)
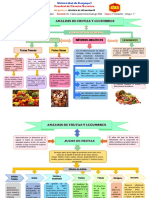
- Analisis de Frutas y LegumbresDocumento5 páginasAnalisis de Frutas y LegumbresGenesis Fajardo IgotsevenAún no hay calificaciones
- HierbabuenaDocumento1 páginaHierbabuenaGenesis Fajardo IgotsevenAún no hay calificaciones
- Guía de Práctica 2Documento14 páginasGuía de Práctica 2Genesis Fajardo IgotsevenAún no hay calificaciones
- Clorofila y Sus TiposDocumento2 páginasClorofila y Sus TiposGenesis Fajardo IgotsevenAún no hay calificaciones
- Celulosa Concepto y EstructuraDocumento8 páginasCelulosa Concepto y EstructuraGenesis Fajardo IgotsevenAún no hay calificaciones
- Obtención de Materia Seca de Cebada, Alfalfa y Chala en Secado Del Ambiente ExternoDocumento9 páginasObtención de Materia Seca de Cebada, Alfalfa y Chala en Secado Del Ambiente ExternoCristobal Severo Huamani CarrionAún no hay calificaciones
- Cómo Se Prepara La Coca ColaDocumento2 páginasCómo Se Prepara La Coca ColaKevin J Crisanto PalaciosAún no hay calificaciones
- Cloruro de TerbutiloDocumento6 páginasCloruro de Terbutiloyocepunkrocker100% (1)
- PROTEÍNAS. García - GutierrezDocumento12 páginasPROTEÍNAS. García - GutierrezAlexx GarcíaRomAún no hay calificaciones
- SolubilidadDocumento13 páginasSolubilidadJhenerson Jhordany Villanueva MedinaAún no hay calificaciones
- Cuáles Son Las Proteínas Presentes en La LecheDocumento12 páginasCuáles Son Las Proteínas Presentes en La LechePaulina Zamudio SarquiAún no hay calificaciones
- Desengomado 4Documento1 páginaDesengomado 4Berenice ChamorroAún no hay calificaciones
- Clase Práctica No.11 Cap. I LípidosDocumento5 páginasClase Práctica No.11 Cap. I LípidosIVANNA FOX BUSTAMANTEAún no hay calificaciones
- Informe - Determinación Del Porcentaje de Acidez Titulable de Una FrutaDocumento6 páginasInforme - Determinación Del Porcentaje de Acidez Titulable de Una FrutaFrias Alfonso SandraAún no hay calificaciones
- Aspectos Teóricos de La Acción de Los FármacosDocumento27 páginasAspectos Teóricos de La Acción de Los FármacosZaida AlvarengaAún no hay calificaciones
- Tesis CatalizadoresDocumento306 páginasTesis CatalizadoresJosé Cortesia100% (1)
- Guia LaboratorioDocumento7 páginasGuia Laboratoriojose contrerasAún no hay calificaciones
- Nano Celulosa - IntroduccionDocumento16 páginasNano Celulosa - IntroduccionAnahiAún no hay calificaciones
- Sem 04 Industria y Lluvia ÁcidaDocumento6 páginasSem 04 Industria y Lluvia ÁcidaAlexander GarciaAún no hay calificaciones
- Proyecto Metabolismo SecundarioDocumento34 páginasProyecto Metabolismo SecundarioJohan BravoAún no hay calificaciones
- Control CV2 RCsDocumento1 páginaControl CV2 RCsAna GilabertAún no hay calificaciones
- Reacciones QuimicasDocumento5 páginasReacciones QuimicasAbimael Para TlvAún no hay calificaciones
- Ada02 Qbio Grupo U1 05Documento14 páginasAda02 Qbio Grupo U1 05Maricarmen Bleis UcAún no hay calificaciones
- Química de La TroposferaDocumento17 páginasQuímica de La Troposferabernardo hurozAún no hay calificaciones
- Metodología EnchiladasDocumento10 páginasMetodología EnchiladasErick Ramírez GonzalezAún no hay calificaciones
- Guía 3 2022 - Licenciatura en ObstetriciaDocumento4 páginasGuía 3 2022 - Licenciatura en ObstetriciaEmmanuel LequiAún no hay calificaciones
- 9 Sintesis de ButanonaDocumento5 páginas9 Sintesis de ButanonaChiquinquira AnguloAún no hay calificaciones
- III Bimestre-BIOLOGÍA-5TO-SECUNDARIADocumento64 páginasIII Bimestre-BIOLOGÍA-5TO-SECUNDARIAMetade Marvel A ChAún no hay calificaciones
- Productos de LimpiezaDocumento6 páginasProductos de Limpiezarafael zavalaAún no hay calificaciones
- Zunchos PetDocumento1 páginaZunchos PetpacevedoAún no hay calificaciones
- Farmacos Moduladores de La AcDocumento35 páginasFarmacos Moduladores de La AcVilma Brizaida Charalla0% (1)
- Hidrocraqueo y Reformado Catalitico PDFDocumento37 páginasHidrocraqueo y Reformado Catalitico PDFsorokzAún no hay calificaciones
- 3 - 2 Estereoisomería de Los MonosacáridosDocumento4 páginas3 - 2 Estereoisomería de Los MonosacáridosMaria Pia Rodriguez SommaAún no hay calificaciones
- Capitulo 2Documento30 páginasCapitulo 2WALDO ADRIAN MAGALLANES PACHASAún no hay calificaciones
- Practica AdnDocumento2 páginasPractica AdnNarukoAún no hay calificaciones