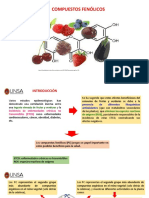
También podría gustarte
- Trabajo de Metabolitos SecundariosDocumento30 páginasTrabajo de Metabolitos SecundariosNatalia NisperuzaAún no hay calificaciones
- FLAVONOIDESDocumento15 páginasFLAVONOIDESFátima FigueroaAún no hay calificaciones
- Productos NaturalesDocumento40 páginasProductos NaturalesEstebanCastellanosBorreroAún no hay calificaciones
- Info de Complejo B RaditininaDocumento22 páginasInfo de Complejo B RaditininaHENRYAún no hay calificaciones
- Clasificación de Flavonoides. (Final)Documento16 páginasClasificación de Flavonoides. (Final)Juan Manuel Sterling100% (1)
- Clasificación de los principales flavonoides: flavanoles, flavanonas, flavonoles, isoflavonas, flavonas y antocianinasDocumento16 páginasClasificación de los principales flavonoides: flavanoles, flavanonas, flavonoles, isoflavonas, flavonas y antocianinasJUAN PABLO CHIQUITO LOAIZAAún no hay calificaciones
- Informe de Bromatologia-Romero Barzola Rebeca GiovanaDocumento56 páginasInforme de Bromatologia-Romero Barzola Rebeca GiovanaAkira Santi100% (1)
- Residuos OrganofosforadosDocumento12 páginasResiduos OrganofosforadosIvan TaboadaAún no hay calificaciones
- PONENCIADocumento7 páginasPONENCIAAngie BedoyaAún no hay calificaciones
- FENOLESDocumento8 páginasFENOLESest.020218230Aún no hay calificaciones
- Trabajo Final - Fase 5 - 301203 - 8Documento26 páginasTrabajo Final - Fase 5 - 301203 - 8Maria Paula Osorio SantosAún no hay calificaciones
- Fenoles, Cresoles y Naftoles. Formulación, usosDocumento14 páginasFenoles, Cresoles y Naftoles. Formulación, usosAlexis LópezAún no hay calificaciones
- Flavonoide PDFDocumento18 páginasFlavonoide PDFRoxana FloresAún no hay calificaciones
- Moléculas: Flavonoides Vegetales: Características Químicas y Actividad BiológicaDocumento16 páginasMoléculas: Flavonoides Vegetales: Características Químicas y Actividad BiológicaCarla Nava MagriAún no hay calificaciones
- Identificación de Compuestos FenolicosDocumento3 páginasIdentificación de Compuestos FenolicosNicolas MacasAún no hay calificaciones
- Características Especializadas Del Metabolismo de Las Plantas e Impacto en La Producción Biotecnológica de Moléculas ObjetivoDocumento18 páginasCaracterísticas Especializadas Del Metabolismo de Las Plantas e Impacto en La Producción Biotecnológica de Moléculas ObjetivoMery Gissela GuamánAún no hay calificaciones
- Guia Lab. Fitoquimica. Identificacion Cualitativa Flavonoides y AlcaloidesDocumento3 páginasGuia Lab. Fitoquimica. Identificacion Cualitativa Flavonoides y AlcaloidesJose David Perez NavarroAún no hay calificaciones
- FLAVONOIDESDocumento11 páginasFLAVONOIDESMónica Del Rio ZuñigaAún no hay calificaciones
- Compuestos FenolicosDocumento12 páginasCompuestos FenolicoserleyramirezAún no hay calificaciones
- Informe de Organica 9 yDocumento6 páginasInforme de Organica 9 ybrayant moralesAún no hay calificaciones
- AzolesDocumento39 páginasAzolesmunozdelgadokarlaAún no hay calificaciones
- Pardeamiento EnzimaticoDocumento13 páginasPardeamiento EnzimaticoPaco OlveraAún no hay calificaciones
- FlavonoideDocumento17 páginasFlavonoideCarl BridgeAún no hay calificaciones
- Polifenoles de Aplicación en Farmacia: Metabolismo y Acción BiológicaDocumento6 páginasPolifenoles de Aplicación en Farmacia: Metabolismo y Acción Biológicajuan montiel bocanellAún no hay calificaciones
- POLIFENOLESDocumento46 páginasPOLIFENOLESRafael Eduardo Gonzalez IbarraAún no hay calificaciones
- FarmacognosiaDocumento23 páginasFarmacognosiaBrenda Alessandra Villanueva ChavezAún no hay calificaciones
- Química Natural y Farmacéutica Grado UAMDocumento10 páginasQuímica Natural y Farmacéutica Grado UAMCarlos Naldo Castillo DelgadoAún no hay calificaciones
- Farmacología CuestionarioDocumento10 páginasFarmacología CuestionarioVicene Norambuena UrrutiaAún no hay calificaciones
- Trabajo de InvestigacionDocumento16 páginasTrabajo de Investigaciondarth_thorionAún no hay calificaciones
- Compuestos fenólicos con actividad anticoagulante de Oenothera roseaDocumento116 páginasCompuestos fenólicos con actividad anticoagulante de Oenothera roseamonicucha1Aún no hay calificaciones
- Determinacion-Polifenoles Totales en QuinuaDocumento8 páginasDeterminacion-Polifenoles Totales en QuinuaRAQUEL NANCY VELIZ SAGARVINAGAAún no hay calificaciones
- Composición nutricional y funcional de algas rojas chilenasDocumento32 páginasComposición nutricional y funcional de algas rojas chilenasCamilo OlavarríaAún no hay calificaciones
- Los Compuestos BioactivosDocumento12 páginasLos Compuestos BioactivosMatii LamasAún no hay calificaciones
- 10 1007@s10532-020-09918-7 en EsDocumento11 páginas10 1007@s10532-020-09918-7 en EsSophie AvilaAún no hay calificaciones
- Metabolismo Secundario de PlantasDocumento27 páginasMetabolismo Secundario de PlantasYue Ichiban-jō No AneAún no hay calificaciones
- Presentación4 COMPUESTOS FENOLICOS PIGMENTOSDocumento38 páginasPresentación4 COMPUESTOS FENOLICOS PIGMENTOSE Clinton LMAún no hay calificaciones
- Policetidos Tipo Macrolido y PolieterDocumento23 páginasPolicetidos Tipo Macrolido y PolieterAlex osorioAún no hay calificaciones
- Aromaticos Policiclicos MarcadoresDocumento14 páginasAromaticos Policiclicos MarcadoresCarlos RenteriaAún no hay calificaciones
- Los Polifenoles de Los Alimentos y La Salud: F. A. Tomás-BarberánDocumento13 páginasLos Polifenoles de Los Alimentos y La Salud: F. A. Tomás-BarberánfloAún no hay calificaciones
- Pec 2 2023 2024Documento4 páginasPec 2 2023 2024AnaAún no hay calificaciones
- Compuestos OxigenadosDocumento9 páginasCompuestos OxigenadosMarcos YaviAún no hay calificaciones
- PR. 9 Determinación de Fenoles Como Agentes Anti-NutricionalesDocumento5 páginasPR. 9 Determinación de Fenoles Como Agentes Anti-NutricionalesLesly Nayeli JPAún no hay calificaciones
- Quercetina PDFDocumento3 páginasQuercetina PDFMarcoantonio MolinagomezAún no hay calificaciones
- Determinacion de PolifenolesDocumento9 páginasDeterminacion de PolifenolesDavid CataguaAún no hay calificaciones
- Dialnet LosCompuestosFenolicosUnAcercamientoASuBiosintesis 6383704 PDFDocumento24 páginasDialnet LosCompuestosFenolicosUnAcercamientoASuBiosintesis 6383704 PDFnancy bautistaAún no hay calificaciones
- Seminario-Semana 9Documento23 páginasSeminario-Semana 9shirleyAún no hay calificaciones
- FurfuralDocumento19 páginasFurfuralFernanda Guadalupe Ramírez MuñozAún no hay calificaciones
- Identificación de Flavonoides en Las PlantasDocumento4 páginasIdentificación de Flavonoides en Las Plantas89jesusAún no hay calificaciones
- Determinación de polifenoles totales por el método de Folin-CiocalteuDocumento9 páginasDeterminación de polifenoles totales por el método de Folin-CiocalteuOmar Gutierrez100% (1)
- Farmacognosia-planta medicinalesDocumento12 páginasFarmacognosia-planta medicinalesYolima Jiménez NausanAún no hay calificaciones
- Guia Lab. Fitoquimica. Identificacion Cualitativa FlavonoidesDocumento3 páginasGuia Lab. Fitoquimica. Identificacion Cualitativa FlavonoidesSAYANDI PAOLA HERNANDEZ CASTELLONAún no hay calificaciones
- POLIFENOLESDocumento9 páginasPOLIFENOLESXiomara Ortega BetancoAún no hay calificaciones
- Fase 4 Bioquimica VegetalDocumento33 páginasFase 4 Bioquimica Vegetaljavier marzan torresAún no hay calificaciones
- Temas selectos de química de productos naturalesDe EverandTemas selectos de química de productos naturalesAún no hay calificaciones
- Química de macrocomponentes de alimentosDe EverandQuímica de macrocomponentes de alimentosAún no hay calificaciones
- Biotecnología enfocada al sector agropecuario y minero con guías de laboratorioDe EverandBiotecnología enfocada al sector agropecuario y minero con guías de laboratorioCalificación: 5 de 5 estrellas5/5 (1)
- Metabolitos secundarios, actividad biológica y etnobotánica de plantas de Santa MartaDe EverandMetabolitos secundarios, actividad biológica y etnobotánica de plantas de Santa MartaAún no hay calificaciones
- Principios De Nutrición De RumiantesDe EverandPrincipios De Nutrición De RumiantesAún no hay calificaciones
- Manual de prácticas avanzadas para el estudio de la Microbiología ambiental de agua y sueloDe EverandManual de prácticas avanzadas para el estudio de la Microbiología ambiental de agua y sueloCalificación: 4 de 5 estrellas4/5 (1)
- InfluenciaDocumento4 páginasInfluenciacreateopAún no hay calificaciones
- Clase D HemocultivosDocumento24 páginasClase D HemocultivosAlfredo Jose Plata RiveraAún no hay calificaciones
- 4-Camaró Sala Et Al 2015Documento6 páginas4-Camaró Sala Et Al 2015Alfredo Jose Plata RiveraAún no hay calificaciones
- Articulo 3 VectoresDocumento39 páginasArticulo 3 VectoresAlfredo Jose Plata RiveraAún no hay calificaciones
- 1 s2.0 S0325754119300811 MainDocumento12 páginas1 s2.0 S0325754119300811 MainIsrael MillanAún no hay calificaciones
- Antagonismo MicrobianoDocumento3 páginasAntagonismo MicrobianoAlfredo Jose Plata RiveraAún no hay calificaciones
- ALFREDO Organismo ModeloDocumento2 páginasALFREDO Organismo ModeloAlfredo Jose Plata RiveraAún no hay calificaciones
- Introduccion Microbiologia VeterinariaDocumento31 páginasIntroduccion Microbiologia VeterinariaAlfredo Jose Plata RiveraAún no hay calificaciones
- Castro Oscar Eq6 P6 PDFDocumento6 páginasCastro Oscar Eq6 P6 PDFAlfredo Jose Plata RiveraAún no hay calificaciones
- Curso Introductorio de Comunicación Científica en Ciencias de La Salud-Certificado Del Curso 2342135Documento1 páginaCurso Introductorio de Comunicación Científica en Ciencias de La Salud-Certificado Del Curso 2342135Alfredo Jose Plata RiveraAún no hay calificaciones
- Taller Educacion para La DemocraciaDocumento2 páginasTaller Educacion para La DemocraciaAlfredo Jose Plata RiveraAún no hay calificaciones
- Reflexion Crecimiento Intragrupal 2Documento2 páginasReflexion Crecimiento Intragrupal 2Alfredo Jose Plata RiveraAún no hay calificaciones
- Microbiología agrícolaDocumento7 páginasMicrobiología agrícolaAlfredo Jose Plata RiveraAún no hay calificaciones
- ENZIMADocumento8 páginasENZIMAAlfredo Jose Plata RiveraAún no hay calificaciones
- Taller BiotecnologíaDocumento3 páginasTaller BiotecnologíaAlfredo Jose Plata RiveraAún no hay calificaciones
- Universidad Simon Bolivar: Fecha de Emision Cuenta NoDocumento1 páginaUniversidad Simon Bolivar: Fecha de Emision Cuenta NoAlfredo Jose Plata RiveraAún no hay calificaciones
- Microbioma HumanoDocumento10 páginasMicrobioma Humanoofe100% (3)
- Trabajo en ClaseDocumento4 páginasTrabajo en ClaseAlfredo Jose Plata RiveraAún no hay calificaciones
- Taller Concepto HHSSDocumento2 páginasTaller Concepto HHSSAlfredo Jose Plata RiveraAún no hay calificaciones
- Taller Parcial Microbiología AgrícolaDocumento7 páginasTaller Parcial Microbiología AgrícolaAlfredo Jose Plata RiveraAún no hay calificaciones
- Autorización Medicamentos Por Utilizar en La IPS: Documento: 1041701492Documento1 páginaAutorización Medicamentos Por Utilizar en La IPS: Documento: 1041701492Alfredo Jose Plata RiveraAún no hay calificaciones
- Linea Del TiempoDocumento1 páginaLinea Del TiempoAlfredo Jose Plata RiveraAún no hay calificaciones
- Taller Parcial Microbiología AgrícolaDocumento6 páginasTaller Parcial Microbiología AgrícolaAlfredo Jose Plata RiveraAún no hay calificaciones
- TallerDocumento2 páginasTallerAlfredo Jose Plata RiveraAún no hay calificaciones
- Reflexion Investigacion Intragrupal Crecimiento Intragrupal 2Documento2 páginasReflexion Investigacion Intragrupal Crecimiento Intragrupal 2Alfredo Jose Plata RiveraAún no hay calificaciones
- 5 - Fe22 - 2021 01 16Documento1 página5 - Fe22 - 2021 01 16Alfredo Jose Plata RiveraAún no hay calificaciones
- Tú Eliges 0% 2021 1 0Documento7 páginasTú Eliges 0% 2021 1 0Alfredo Jose Plata RiveraAún no hay calificaciones
- View ReportDocumento1 páginaView ReportAlfredo Jose Plata RiveraAún no hay calificaciones
- Reflexion Crecimiento Intragrupal 2Documento2 páginasReflexion Crecimiento Intragrupal 2Alfredo Jose Plata RiveraAún no hay calificaciones
- Helados y MantequillaDocumento35 páginasHelados y MantequillaAndres Felipe Bolaño NarvaezAún no hay calificaciones
- Rol Microorganismos en Ind. AlimentariaDocumento13 páginasRol Microorganismos en Ind. Alimentarialeslie selene100% (1)
- Quesos Cottage y Tipo MozarellaDocumento2 páginasQuesos Cottage y Tipo MozarellaAlberto AyónAún no hay calificaciones
- Leche Acidofilas y FermentadasDocumento42 páginasLeche Acidofilas y Fermentadasfarina moretiAún no hay calificaciones
- Vida Util de Carne Fresca de Res Envasada Al VacioDocumento19 páginasVida Util de Carne Fresca de Res Envasada Al VacioLuis Carlos V. RamirezAún no hay calificaciones
- Efecto Del Crecimiento Microbiano Sobre Las Propiedades de La CarneDocumento7 páginasEfecto Del Crecimiento Microbiano Sobre Las Propiedades de La CarneDiego Raul Puche OrtizAún no hay calificaciones
- Yogurt RecursamientoDocumento12 páginasYogurt RecursamientoJurassicworldgodzillapokemon Mata maestrosAún no hay calificaciones
- Tecnología del salado y salmuera en quesosDocumento53 páginasTecnología del salado y salmuera en quesosmeryAún no hay calificaciones
- Leche FermentadaDocumento8 páginasLeche FermentadaMARIA LORENSA DIAZ MOLINAAún no hay calificaciones
- Rseña Perfil Microbiológico de La Harina de Maíz Nixtamalizada FermentadaDocumento1 páginaRseña Perfil Microbiológico de La Harina de Maíz Nixtamalizada FermentadaJonathan MarquezAún no hay calificaciones
- Leche Clase IIDocumento38 páginasLeche Clase IIirma cerna vasquezAún no hay calificaciones
- Clase 5. Tecnologia Quesera-1Documento52 páginasClase 5. Tecnologia Quesera-1Jaqueline Edith Panibra MontoyaAún no hay calificaciones
- Trabajo de Investigacion - Fermentacion de AlimentosDocumento34 páginasTrabajo de Investigacion - Fermentacion de AlimentosAlysson MarlethAún no hay calificaciones
- Taller 3Documento19 páginasTaller 3jorge severinoAún no hay calificaciones
- Frutas y HortalizasDocumento17 páginasFrutas y HortalizaskellyAún no hay calificaciones
- Biotecnología y AlimentaciónDocumento376 páginasBiotecnología y AlimentaciónJacinto Luque Aguilar100% (1)
- El Portillo LtdaDocumento22 páginasEl Portillo LtdaMariale Africano Barrera100% (1)
- Cuestionario Final MicroDocumento13 páginasCuestionario Final MicroKatherinne BorjaAún no hay calificaciones
- Tecnologia para La Elaboracion de QuesoDocumento16 páginasTecnologia para La Elaboracion de Quesoand3sballesAún no hay calificaciones
- Leches Fermentadas o AcidasDocumento12 páginasLeches Fermentadas o AcidasElkin MirandaAún no hay calificaciones
- MICROBIOLOGÍA DE LA LECHE ResumenDocumento7 páginasMICROBIOLOGÍA DE LA LECHE Resumenanon_963218119Aún no hay calificaciones
- NOTA: No Trabajar Desde Drive, Descargar La Plantilla (Archivo / Descargar) - en Este Video Se Muestra Cómo HacerloDocumento10 páginasNOTA: No Trabajar Desde Drive, Descargar La Plantilla (Archivo / Descargar) - en Este Video Se Muestra Cómo HacerloAbi GarciaAún no hay calificaciones
- Produccion de Acido LácticoDocumento60 páginasProduccion de Acido LácticoMiguel FerrerAún no hay calificaciones
- Metabolismo microbiano: respiración y fermentaciónDocumento18 páginasMetabolismo microbiano: respiración y fermentaciónRicardoAún no hay calificaciones
- I. IntroducciónDocumento112 páginasI. IntroducciónMarialy AlvarezAún no hay calificaciones
- Equipo D - Trabajo EncargadoDocumento21 páginasEquipo D - Trabajo EncargadoKusar Avendaño CaballeroAún no hay calificaciones
- NORMA Oficial Mexicana NOM-051-SCFI - SSA1-2010, Especificaciones Generales de Etiquetado para Alimentos y Bebidas No Alcohólicas Preenvasados-Información Comercial y SanitariaDocumento21 páginasNORMA Oficial Mexicana NOM-051-SCFI - SSA1-2010, Especificaciones Generales de Etiquetado para Alimentos y Bebidas No Alcohólicas Preenvasados-Información Comercial y SanitariaMartha NavarreteAún no hay calificaciones
- Lactobacillus, Streptococos y clasificación del yogurtDocumento17 páginasLactobacillus, Streptococos y clasificación del yogurtLizzeth RojasPAún no hay calificaciones
- Cultivos ForrajerosDocumento9 páginasCultivos ForrajerosERIKA MAGDALENA ANDRADE ANDRADEAún no hay calificaciones