También podría gustarte
- Inductores EndovenososDocumento46 páginasInductores EndovenososSAMUEL MARIN FUQUENEAún no hay calificaciones
- Taller 3 - Anestésicos IntravenososDocumento9 páginasTaller 3 - Anestésicos Intravenososjuan andres lopezAún no hay calificaciones
- IntubaciónDocumento25 páginasIntubaciónMaria Isabel CeballosAún no hay calificaciones
- Fichas FarmacológicasDocumento17 páginasFichas FarmacológicasKaren Manayalle CespedesAún no hay calificaciones
- AnticonvulsivantesDocumento66 páginasAnticonvulsivantesJad Steppenwolf100% (2)
- Anestésicos Generales y LocalesDocumento6 páginasAnestésicos Generales y LocalesYang ChichoAún no hay calificaciones
- FARMACO1Documento26 páginasFARMACO1Robert JimenezAún no hay calificaciones
- InductoresDocumento56 páginasInductoresNayeli GarcíaAún no hay calificaciones
- Expo InductoresDocumento8 páginasExpo InductoresDiana GomezAún no hay calificaciones
- 5-Hta, Purinas, Neuropéptidos y CannabinoidesDocumento6 páginas5-Hta, Purinas, Neuropéptidos y CannabinoidesRAUL STEVEN SEPULVEDA GONZALEZAún no hay calificaciones
- Fármacos Antihistamínicos y Serotoninérgicos IIIDocumento12 páginasFármacos Antihistamínicos y Serotoninérgicos IIIlalalupsi12Aún no hay calificaciones
- Tabla de Fármacos S6Documento14 páginasTabla de Fármacos S6CARLOS MANUEL MARI�OS CARRANZAAún no hay calificaciones
- No BarbituricosDocumento40 páginasNo BarbituricosMaria Luisa Mercedez MonegroAún no hay calificaciones
- Ilovepdf MergedDocumento174 páginasIlovepdf MergedGISSELY CRISPINA BENANCIO RODRIGUEZAún no hay calificaciones
- TRANQUILIZANTESDocumento63 páginasTRANQUILIZANTESCarolina ÁlvarezAún no hay calificaciones
- Clase 3 - Farmacologia Del SNCDocumento35 páginasClase 3 - Farmacologia Del SNCALVARO JESUS GUTIERREZ TOLEDOAún no hay calificaciones
- Agonistas y Antagonistas Del S. N ParasimpaticoDocumento28 páginasAgonistas y Antagonistas Del S. N ParasimpaticoDavis alberto Mejia pinedoAún no hay calificaciones
- Anestesicos Generales y LocalesDocumento6 páginasAnestesicos Generales y LocalesAlvaro MedinaAún no hay calificaciones
- Informe Practica N 8Documento15 páginasInforme Practica N 8ELIAún no hay calificaciones
- AnsioliticosDocumento10 páginasAnsioliticosJP KantAún no hay calificaciones
- Fármacos Ansiolíticos e HipnóticosDocumento5 páginasFármacos Ansiolíticos e HipnóticosJuan OrellanaAún no hay calificaciones
- Informe de Práctica 1 FármacoDocumento26 páginasInforme de Práctica 1 FármacocarlosAún no hay calificaciones
- BENZODIACEPINASDocumento25 páginasBENZODIACEPINASSophie DíazAún no hay calificaciones
- Farmacos en AnestesiaDocumento62 páginasFarmacos en AnestesiaelizabethAún no hay calificaciones
- Intoxicación POR PSICOFÁRMACOS y AlcoholesDocumento122 páginasIntoxicación POR PSICOFÁRMACOS y AlcoholesRudy FukudaAún no hay calificaciones
- AntiepilepticosDocumento20 páginasAntiepilepticosEmiliano CatanaAún no hay calificaciones
- Fármacos AntiepilépticosDocumento8 páginasFármacos AntiepilépticosculosoAún no hay calificaciones
- AnticonvulsivantesDocumento14 páginasAnticonvulsivantesIsa Vasquez PAún no hay calificaciones
- Relajantes MuscularesDocumento19 páginasRelajantes MuscularesBrianna Salazar100% (1)
- 5hta, Purinas, Neuropeptidos y CannabinoidesDocumento28 páginas5hta, Purinas, Neuropeptidos y CannabinoidesLety LagunaAún no hay calificaciones
- Ansioliticos e HipnoticosDocumento73 páginasAnsioliticos e HipnoticosGian Franco Antonio RubioAún no hay calificaciones
- Enfermedad de Alzheimer ExposicionDocumento126 páginasEnfermedad de Alzheimer ExposicionJhonatan Tuñoque DíazAún no hay calificaciones
- AntidepresivosDocumento55 páginasAntidepresivosf.torres14Aún no hay calificaciones
- PisadolDocumento4 páginasPisadolmvz.r.a.pAún no hay calificaciones
- Farmacos para Cirugia VeterinariaDocumento6 páginasFarmacos para Cirugia VeterinariaJim Gilbert0% (1)
- Medicamentos para El Síndrome DolorosoDocumento7 páginasMedicamentos para El Síndrome DolorosoMARGHEL MADAIZ ALVARADO SALASAún no hay calificaciones
- Farmacología de Los Agentes AnestésicosDocumento22 páginasFarmacología de Los Agentes AnestésicosNinfa RomeroAún no hay calificaciones
- Anestésicos LocalesDocumento35 páginasAnestésicos LocalesMarissa JiménezAún no hay calificaciones
- Antagonistas ColinergicosDocumento48 páginasAntagonistas ColinergicosAnonymous JuJACxjAún no hay calificaciones
- AntidepresivosDocumento34 páginasAntidepresivosMaida TorresAún no hay calificaciones
- Anestesicos y AnalgesicoDocumento10 páginasAnestesicos y Analgesicoluisdaniel molinaAún no hay calificaciones
- Clase de Antimigrañosos y AntigotososDocumento59 páginasClase de Antimigrañosos y AntigotososCynthia Gallo100% (1)
- Fármacos Antiepilépticos y AnticonvulsivantesDocumento47 páginasFármacos Antiepilépticos y AnticonvulsivantesKEVIN BRACHIEL BAZAN SEGURAAún no hay calificaciones
- Farmacología Del SNC: MG Elizabeth Hinostroza MezaDocumento49 páginasFarmacología Del SNC: MG Elizabeth Hinostroza MezaJoana TelloAún no hay calificaciones
- Fármacos Antiadrenérgicos - Efectos Sistémicos, Usos Terapéuticos (Q.F.Pablo Iturra)Documento35 páginasFármacos Antiadrenérgicos - Efectos Sistémicos, Usos Terapéuticos (Q.F.Pablo Iturra)mauro soudrecamposAún no hay calificaciones
- Semanal AnestesioDocumento57 páginasSemanal AnestesioJhommy ChimborazoAún no hay calificaciones
- Anestésicos Ev e InhalatoriosDocumento29 páginasAnestésicos Ev e Inhalatoriosdm.impresionesAún no hay calificaciones
- Inductores AnestesicosDocumento79 páginasInductores AnestesicosAna Luisa PAún no hay calificaciones
- Vías de Señalización de ReceptoresDocumento14 páginasVías de Señalización de ReceptoresKEREN VIVIANA GASCON YEPESAún no hay calificaciones
- Receptores y Neurotransmisores - LJFRDocumento131 páginasReceptores y Neurotransmisores - LJFRcamila freyreAún no hay calificaciones
- Tiva 2023Documento104 páginasTiva 2023Noel MallaAún no hay calificaciones
- Neurode Pre SoresDocumento34 páginasNeurode Pre Soreskarlos0407Aún no hay calificaciones
- Fármacos Antihistamínicos y Serotoninérgicos II (Clase 17)Documento15 páginasFármacos Antihistamínicos y Serotoninérgicos II (Clase 17)lalalupsi12Aún no hay calificaciones
- AntimuscarínicosDocumento28 páginasAntimuscarínicosSheyla CoursonAún no hay calificaciones
- Previo 1. Ansioliticos y DiagDocumento7 páginasPrevio 1. Ansioliticos y DiagFosado AleAún no hay calificaciones
- Intoxicacion Por BenzodiacepinaDocumento35 páginasIntoxicacion Por BenzodiacepinaElkin Niño Jara0% (1)
- Practica 7 y 8Documento22 páginasPractica 7 y 8Wilma Valiente TantaAún no hay calificaciones
- Broncodilatadores: - Pablo Campos L Magister en EducaciónDocumento25 páginasBroncodilatadores: - Pablo Campos L Magister en EducaciónValeria NavarroAún no hay calificaciones
- Relajantes Musculares y ParkinsonDocumento8 páginasRelajantes Musculares y ParkinsonIsa Vasquez PAún no hay calificaciones
- Aprenda Farmacología En Una SemanaDe EverandAprenda Farmacología En Una SemanaCalificación: 2 de 5 estrellas2/5 (1)
- Infrograma Sindrome de DownDocumento1 páginaInfrograma Sindrome de Downj100% (1)
- Psa en MujeresDocumento1 páginaPsa en MujeresAsesoria TecnicaAún no hay calificaciones
- Grupo 1 Modelos de CausalidadDocumento12 páginasGrupo 1 Modelos de CausalidadJosselyn Adriana Becerra VelezAún no hay calificaciones
- Producción de Antibioticos A Escala Industrial - E2Documento47 páginasProducción de Antibioticos A Escala Industrial - E2Cruz Jiménez Katia SeleneAún no hay calificaciones
- Artrosis Expo 03Documento10 páginasArtrosis Expo 03Diana Polo VeraAún no hay calificaciones
- Radiología e ImagenologiaDocumento6 páginasRadiología e ImagenologiaVictor Oswaldo Martínez AguAún no hay calificaciones
- Respuestas Cardiovasculares Al EjercicioDocumento10 páginasRespuestas Cardiovasculares Al EjercicioJose Ivan MantillaAún no hay calificaciones
- Apendicitis AgudaDocumento18 páginasApendicitis AgudaSmirf FeimsAún no hay calificaciones
- Natalia Gonzales de Prada - PORTAFOLIO Hito 3 ProcesualDocumento8 páginasNatalia Gonzales de Prada - PORTAFOLIO Hito 3 ProcesualNatalia PradaAún no hay calificaciones
- Geriátrico: Educación SanitariaDocumento6 páginasGeriátrico: Educación SanitariaJaime Pérez CabreraAún no hay calificaciones
- Patron IntersticialDocumento9 páginasPatron IntersticialEsthefanyAún no hay calificaciones
- Kalichman 2011Documento13 páginasKalichman 2011Anabella ScottiAún no hay calificaciones
- Patología Benigna y Maligna de HígadoDocumento12 páginasPatología Benigna y Maligna de HígadoLeon Jilo0% (1)
- GRUPO7 - Aneurisma Cerebral y Hemorragia Subaracnoidea-PAEDocumento9 páginasGRUPO7 - Aneurisma Cerebral y Hemorragia Subaracnoidea-PAEStefania ZambranoAún no hay calificaciones
- 2.clasificacion de Las Enfermedades AutoinmunesDocumento20 páginas2.clasificacion de Las Enfermedades AutoinmunesMarcelo BecksAún no hay calificaciones
- TRIPTICO HGW PERU ModificadoDocumento3 páginasTRIPTICO HGW PERU ModificadoRonald inga julca67% (3)
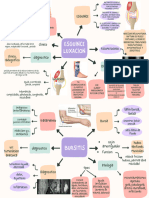
- Bursitis, Luxacion y EsguinseDocumento2 páginasBursitis, Luxacion y Esguinseazucena lopezAún no hay calificaciones
- Silabo 2017 IIDocumento8 páginasSilabo 2017 IIHector Chumpitaz VelasquezAún no hay calificaciones
- Patologias en EquinosDocumento26 páginasPatologias en EquinosKelita Orellana100% (4)
- Tesis Analisis Del Proyecto de Ley Que Regula Los Principios Jurudicos - Image.markedDocumento250 páginasTesis Analisis Del Proyecto de Ley Que Regula Los Principios Jurudicos - Image.markedRaúl S. RodríguezAún no hay calificaciones
- PENFIGODocumento5 páginasPENFIGOzara galiciaAún no hay calificaciones
- Parches de DuodermDocumento6 páginasParches de DuodermKevin TzumAún no hay calificaciones
- Resfrio Comun y Gripe-2Documento15 páginasResfrio Comun y Gripe-2ELIAS CISNEROSAún no hay calificaciones
- Notas de Ingreso A Área de ContingenciaDocumento34 páginasNotas de Ingreso A Área de ContingenciaXime MtzAún no hay calificaciones
- PASTA DE LASSAR - Uribe Franco GalileaDocumento9 páginasPASTA DE LASSAR - Uribe Franco GalileaEmmanuel SerranoAún no hay calificaciones
- Clasificación NOCDocumento19 páginasClasificación NOCelsy del rocio z. r.Aún no hay calificaciones
- Mapa Conceptual Historia ClinicaDocumento1 páginaMapa Conceptual Historia Clinicanayhel guevara rojas86% (7)
- Concientizacion en Uso de AntibioticosDocumento5 páginasConcientizacion en Uso de Antibioticoscristopher martinezAún no hay calificaciones
- Curso de Activación y Readaptación Psicomotriz en Geriatría Oct 2023Documento8 páginasCurso de Activación y Readaptación Psicomotriz en Geriatría Oct 2023Cristina RodriguezAún no hay calificaciones
- Nutricion InfantilDocumento13 páginasNutricion InfantilRoDy ViviAún no hay calificaciones