También podría gustarte
- Qué es la Epilepsia. Causas, Síntomas, Diagnóstico y Tratamiento: TRASTORNOS DEL SUEÑO, #2De EverandQué es la Epilepsia. Causas, Síntomas, Diagnóstico y Tratamiento: TRASTORNOS DEL SUEÑO, #2Aún no hay calificaciones
- AnemiasDocumento24 páginasAnemiasluis sulbaranAún no hay calificaciones
- Hepatoesplenomegalia - P1Documento46 páginasHepatoesplenomegalia - P1Gabriela MoralesAún no hay calificaciones
- Asfixia PerinatalDocumento17 páginasAsfixia PerinatalValeria AliagaAún no hay calificaciones
- Reporte de Caso Clínico de Un Paciente Con Leucemia Mieloide CrónicaDocumento12 páginasReporte de Caso Clínico de Un Paciente Con Leucemia Mieloide CrónicaVanesa Rubí CamposAún no hay calificaciones
- Sindromes Meningeo y CerebelosoDocumento18 páginasSindromes Meningeo y CerebelosoRuth Cobo Rosales100% (2)
- Trastornos Hipertensivos Del Embarazo DR PaucarDocumento144 páginasTrastornos Hipertensivos Del Embarazo DR PaucarValia ChuchonAún no hay calificaciones
- Caso Clinico Rotacion NeurologiaDocumento18 páginasCaso Clinico Rotacion NeurologiaPaola N. Rangel TeranAún no hay calificaciones
- Meningitis BacterianaDocumento29 páginasMeningitis BacterianaKenia Zurita ContrerasAún no hay calificaciones
- PREECLAMPSIA ExpoDocumento40 páginasPREECLAMPSIA ExpoLisseth Becerra VargasAún no hay calificaciones
- Sindrome de TurnerDocumento18 páginasSindrome de Turnerchabely2106Aún no hay calificaciones
- CLASE Semana 4. Anemias RegenerativasDocumento56 páginasCLASE Semana 4. Anemias RegenerativasAngelina HernándezAún no hay calificaciones
- Clase Clinica Medica I 20.07.2023 Laboratorio, Enfermedades Infecciosas y Parasitarias. Clinica Medica IDocumento49 páginasClase Clinica Medica I 20.07.2023 Laboratorio, Enfermedades Infecciosas y Parasitarias. Clinica Medica Ifrancis gutembergAún no hay calificaciones
- BAZO 2022 AgtoDocumento18 páginasBAZO 2022 AgtoReina ValenciaAún no hay calificaciones
- Patología EsplénicaDocumento36 páginasPatología EsplénicaJack Edward Peña SolanoAún no hay calificaciones
- Esplenopatías Quirúrgicas No TraumáticasDocumento42 páginasEsplenopatías Quirúrgicas No TraumáticasDaritoAún no hay calificaciones
- Gaucher IDocumento18 páginasGaucher IGabriela Paladines GallegosAún no hay calificaciones
- INSUFICIENCIA HEPÁTICA Antonio Leobardo Ruiz EscobarDocumento40 páginasINSUFICIENCIA HEPÁTICA Antonio Leobardo Ruiz EscobarLeo Ruiz EscobarAún no hay calificaciones
- Insuficiencia HepaticaDocumento42 páginasInsuficiencia HepaticaAriana N. Castillo H.100% (1)
- AMILOIDOSISDocumento9 páginasAMILOIDOSISRocio SanchezAún no hay calificaciones
- Anemia PerniciosaDocumento10 páginasAnemia PerniciosaKarla LizbethAún no hay calificaciones
- Ictericia NeonatalDocumento45 páginasIctericia Neonatalmaria gomez yoveraAún no hay calificaciones
- Sindrome de TurnerDocumento18 páginasSindrome de Turnerchabely2106Aún no hay calificaciones
- NECROSISDocumento48 páginasNECROSISEllen LópezAún no hay calificaciones
- Anemiaa PlasicaDocumento37 páginasAnemiaa PlasicaRael Cordoba RamirezAún no hay calificaciones
- Mega SwagDocumento24 páginasMega SwagPaulo ContadorAún no hay calificaciones
- SEMIOLOGÍA TIROIDEA 1noDocumento45 páginasSEMIOLOGÍA TIROIDEA 1noEver Andres Zegarra CandiottiAún no hay calificaciones
- Anemiaa PlasicaDocumento37 páginasAnemiaa Plasicajulio cesarAún no hay calificaciones
- EndocarditisDocumento5 páginasEndocarditisComision A Comision AAún no hay calificaciones
- Presentacion NefroDocumento35 páginasPresentacion NefroAlejandra PereiraAún no hay calificaciones
- Sindromes Geneticos Mas ComunesDocumento47 páginasSindromes Geneticos Mas ComunesSary Contreras JAún no hay calificaciones
- SIGNOS DE ALARMA DEL RECIEN NACIDO (Recuperado)Documento23 páginasSIGNOS DE ALARMA DEL RECIEN NACIDO (Recuperado)YURIKO VEGA SUAREZ100% (1)
- EsplenomegaliasDocumento41 páginasEsplenomegaliasIsmael Pacheco vaqueroAún no hay calificaciones
- Leucemias 1Documento17 páginasLeucemias 1maria madridAún no hay calificaciones
- Lupus Eritematoso Sistemico PDFDocumento7 páginasLupus Eritematoso Sistemico PDFenani_20Aún no hay calificaciones
- Vol16!1!05 Habilidades e Terapeutica HigadoDocumento42 páginasVol16!1!05 Habilidades e Terapeutica HigadoMarcoAún no hay calificaciones
- Leucemia Granulocítica CronicaDocumento12 páginasLeucemia Granulocítica CronicaGabriel HernándezAún no hay calificaciones
- Pcte AlamosDocumento37 páginasPcte AlamosDaniela CalleAún no hay calificaciones
- Patologías Genéticas GuíaDocumento13 páginasPatologías Genéticas Guíaasofto26Aún no hay calificaciones
- enfermeDAD HEPATICA EN EL EMBARAZODocumento45 páginasenfermeDAD HEPATICA EN EL EMBARAZOPerla PradoAún no hay calificaciones
- Sindrome Nefrotico 14Documento80 páginasSindrome Nefrotico 14paola bendezu saraviaAún no hay calificaciones
- Errores Innatos Del MetabolismoDocumento22 páginasErrores Innatos Del MetabolismoPaola ZanabriaAún no hay calificaciones
- Conceptos Clave PediatríaDocumento11 páginasConceptos Clave Pediatríarami avellanedaAún no hay calificaciones
- Sesión 9 Hipofunción Hipotálamo Hipófisis Necrosis Pituitaria Diabetes InsipidaDocumento44 páginasSesión 9 Hipofunción Hipotálamo Hipófisis Necrosis Pituitaria Diabetes InsipidaKaren Chavez MartinezAún no hay calificaciones
- Guillain BarreDocumento53 páginasGuillain Barrecristian_mg10Aún no hay calificaciones
- Complicaciones Cronicas de DM-23Documento74 páginasComplicaciones Cronicas de DM-23chloenanutAún no hay calificaciones
- Almacenamiento de GlucogenoDocumento13 páginasAlmacenamiento de Glucogenosarith ibarraAún no hay calificaciones
- Linfoma H - NHDocumento18 páginasLinfoma H - NHTracy GamarraAún no hay calificaciones
- Síndrome Antifosfolipídic O: Dra. Cecibel Salamea Sarmiento. Especialista ReumatologíaDocumento31 páginasSíndrome Antifosfolipídic O: Dra. Cecibel Salamea Sarmiento. Especialista Reumatologíanicolas lopezAún no hay calificaciones
- 12 Ictericia NeonatalDocumento77 páginas12 Ictericia NeonatalsasdafafAún no hay calificaciones
- Hematologia CuestionarioDocumento16 páginasHematologia Cuestionarioarios_409594Aún no hay calificaciones
- Asfixia NeonatalDocumento15 páginasAsfixia NeonatalMarina Tlv XzAún no hay calificaciones
- Polocitemias y PoliglobuliasDocumento5 páginasPolocitemias y PoliglobuliasKim FernándezAún no hay calificaciones
- Caso ClinicoDocumento28 páginasCaso ClinicoGris ERAún no hay calificaciones
- Habitus Exterior, Signos Vitales y SomatometríaDocumento42 páginasHabitus Exterior, Signos Vitales y SomatometríaAngel Joaquín MartínezAún no hay calificaciones
- Caso ClinicoDocumento13 páginasCaso ClinicoLEYLA LUZ ESTRELLA BARNARD LAGUNAAún no hay calificaciones
- Convulsiones en El Recien Nacido 2Documento18 páginasConvulsiones en El Recien Nacido 2Anonymous tzu6D0biyfAún no hay calificaciones
- Asfixia PerinatalDocumento25 páginasAsfixia PerinatalAnahi NavarroAún no hay calificaciones
- Sesion 8Documento33 páginasSesion 8Briggith Guayan lavadoAún no hay calificaciones
- Esferocitosis HereditariaDocumento24 páginasEsferocitosis HereditariaGina Melisa UrquizaAún no hay calificaciones
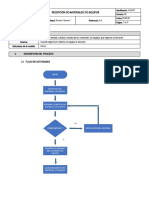
- Alm.i01 Recepción de Materiales y - o EquiposDocumento4 páginasAlm.i01 Recepción de Materiales y - o EquiposjuanpabloveeyAún no hay calificaciones
- Calentadores de Agua de Alimentacion y CondensadoresDocumento36 páginasCalentadores de Agua de Alimentacion y CondensadoresGabrielAún no hay calificaciones
- Listado de Cursos 22-23Documento4 páginasListado de Cursos 22-23harveyal_apAún no hay calificaciones
- Lectura 1Documento7 páginasLectura 1Norelly OlivaresAún no hay calificaciones
- Listado de Clinicas ConvenioDocumento42 páginasListado de Clinicas ConvenioCharly Romero Suarez100% (1)
- Tutoria MSDocumento4 páginasTutoria MSCamila CuzcoAún no hay calificaciones
- Resumen DiafanizacionDocumento1 páginaResumen DiafanizacionjdcolqueAún no hay calificaciones
- E1 EstadisticaDocumento4 páginasE1 EstadisticaUriel Rubio RiveraAún no hay calificaciones
- Riegos AsfálticosDocumento10 páginasRiegos AsfálticosPablo CorreaAún no hay calificaciones
- Extrusora Al Vacio MVB 320Documento3 páginasExtrusora Al Vacio MVB 320Jhonn GonzalesAún no hay calificaciones
- El Reciclaje en El Municipo de MirandaDocumento15 páginasEl Reciclaje en El Municipo de MirandaZanyely de la CruzAún no hay calificaciones
- TAI138Documento98 páginasTAI138Belen intriagoAún no hay calificaciones
- InmunohematologiaDocumento57 páginasInmunohematologiamariatrivilo18504Aún no hay calificaciones
- APA 1 3 Biopsia IntraoperatoriaDocumento6 páginasAPA 1 3 Biopsia IntraoperatoriaJavier HallerAún no hay calificaciones
- Brochure Curso Especializado InocuidadDocumento6 páginasBrochure Curso Especializado InocuidadmfiestasAún no hay calificaciones
- Cartilla Medica Federada SaludDocumento11 páginasCartilla Medica Federada Saludeptaservice34Aún no hay calificaciones
- PDF Practica Determinacion Del Contenido de Saponinas en La Quinua - CompressDocumento18 páginasPDF Practica Determinacion Del Contenido de Saponinas en La Quinua - CompressSantiago DangerAún no hay calificaciones
- Tipificación de AcerosDocumento40 páginasTipificación de AcerosJuan Pablo GutiérrezAún no hay calificaciones
- GRUPO 04 Caso Clínico AstridDocumento28 páginasGRUPO 04 Caso Clínico AstridAstrid MendozaAún no hay calificaciones
- Clase Gestion de La Convivencia Escolar 1Documento64 páginasClase Gestion de La Convivencia Escolar 1PatriciaAún no hay calificaciones
- Trabajo Investigacion Espesantes para Estampado TextilDocumento12 páginasTrabajo Investigacion Espesantes para Estampado TextilÁngela Mione Cultraro0% (1)
- SALUD INTEGRAL ADOLS III - PDFDocumento9 páginasSALUD INTEGRAL ADOLS III - PDFJOSÉ MARTÍNEZ ARELLANOAún no hay calificaciones
- LegislacionDocumento10 páginasLegislacionWanda Batres CórdovaAún no hay calificaciones
- Tarea ELDocumento4 páginasTarea ELBelis Gonzalez Cogollo25% (4)
- Transmisiones de Banda Plana o RedondaDocumento38 páginasTransmisiones de Banda Plana o RedondaOMAR ALEJANDRO LONGORIA VAZQUEZAún no hay calificaciones
- El Viaje Del Alcohol Por El CuerpoDocumento4 páginasEl Viaje Del Alcohol Por El CuerpoWinston SaucedoAún no hay calificaciones
- UNE-EN 13160-5 (Sistemas de Detección de Fugas)Documento86 páginasUNE-EN 13160-5 (Sistemas de Detección de Fugas)Pablo Ibarra Quintana100% (2)
- Tarea - Situación de Salud InfantilDocumento3 páginasTarea - Situación de Salud InfantilCARRASCO G�LVEZ VICTOR DANIELAún no hay calificaciones
- Esquema EndoDocumento1 páginaEsquema EndoRandy LucasAún no hay calificaciones
- Ensayo de La PelículaDocumento4 páginasEnsayo de La PelículaYamile Cetina CaheroAún no hay calificaciones
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Principios de medicina tradicional chinaDe EverandPrincipios de medicina tradicional chinaCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Pare La Diabetes en 14 Dias: No Ataque la Consecuencia de la Diabetes. Ataque la Causa de la DiabetesDe EverandPare La Diabetes en 14 Dias: No Ataque la Consecuencia de la Diabetes. Ataque la Causa de la DiabetesCalificación: 4.5 de 5 estrellas4.5/5 (55)
- El Libro de la Dieta Antiinflamatoria: Plan de 14 días para Sanar el Sistema inmunológico y Sentirte Mejor que NuncaDe EverandEl Libro de la Dieta Antiinflamatoria: Plan de 14 días para Sanar el Sistema inmunológico y Sentirte Mejor que NuncaCalificación: 4.5 de 5 estrellas4.5/5 (14)
- El concepto Mulligan de terapia manual (Color)De EverandEl concepto Mulligan de terapia manual (Color)Calificación: 5 de 5 estrellas5/5 (3)
- Manual de ortopedia maxilar: Modelo diagnóstico de maloclusiones para pacientes en crecimientoDe EverandManual de ortopedia maxilar: Modelo diagnóstico de maloclusiones para pacientes en crecimientoCalificación: 4.5 de 5 estrellas4.5/5 (14)
- Alimentación prebiótica: Para una microbiota intestinal sanaDe EverandAlimentación prebiótica: Para una microbiota intestinal sanaCalificación: 4 de 5 estrellas4/5 (14)
- Fundamentos de medicina tradicional chinaDe EverandFundamentos de medicina tradicional chinaCalificación: 4.5 de 5 estrellas4.5/5 (5)
- Aprendizaje del examen clínico de los equinos, bovinos y caninosDe EverandAprendizaje del examen clínico de los equinos, bovinos y caninosCalificación: 4.5 de 5 estrellas4.5/5 (3)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)
- La anatomía de las lesiones deportivas (Color)De EverandLa anatomía de las lesiones deportivas (Color)Calificación: 4.5 de 5 estrellas4.5/5 (12)
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- El código de la obesidad: Descifrando los secretos de la pérdida de pesoDe EverandEl código de la obesidad: Descifrando los secretos de la pérdida de pesoCalificación: 4.5 de 5 estrellas4.5/5 (51)
- Limpiar, Nutrir, Reparar: Adiós a Las Enfermedades, En Tres Pasos NaturalesDe EverandLimpiar, Nutrir, Reparar: Adiós a Las Enfermedades, En Tres Pasos NaturalesCalificación: 4 de 5 estrellas4/5 (6)
- Cuerpo Tóxico: Como Liberar Tu Cuerpo De Las Toxinas Externas E Internas, Y Evitar Asi Los Efectos De Los Radicales LibresDe EverandCuerpo Tóxico: Como Liberar Tu Cuerpo De Las Toxinas Externas E Internas, Y Evitar Asi Los Efectos De Los Radicales LibresCalificación: 5 de 5 estrellas5/5 (2)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- Cambiar el pasado: Superar las experiencias traumáticas con la terapia estratégicaDe EverandCambiar el pasado: Superar las experiencias traumáticas con la terapia estratégicaCalificación: 5 de 5 estrellas5/5 (4)
- Hierbas Medicinales: La Guía Definitiva para Lograr una Salud ExtraordinariaDe EverandHierbas Medicinales: La Guía Definitiva para Lograr una Salud ExtraordinariaCalificación: 5 de 5 estrellas5/5 (2)
- Gua Sha: Guía de autotratamiento completoDe EverandGua Sha: Guía de autotratamiento completoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- La rueda medicinal del ayurveda: Máxima salud y energía para tu cuerpo, mente y espírituDe EverandLa rueda medicinal del ayurveda: Máxima salud y energía para tu cuerpo, mente y espírituCalificación: 4.5 de 5 estrellas4.5/5 (10)
- Flores de Bach Recursos y estrategias terapéuticasDe EverandFlores de Bach Recursos y estrategias terapéuticasCalificación: 3.5 de 5 estrellas3.5/5 (3)