También podría gustarte
- BF TALLER 7 Diseño de Primers PDFDocumento12 páginasBF TALLER 7 Diseño de Primers PDFIsaac David Abad MatiasAún no hay calificaciones
- PEC - 2 BiologíaDocumento13 páginasPEC - 2 BiologíaJose Miguel CayuelaAún no hay calificaciones
- Guia Practica Sobre La Tecnica PCRDocumento24 páginasGuia Practica Sobre La Tecnica PCRDaniel RomeroAún no hay calificaciones
- Informe Laboratorio 8910-2Documento39 páginasInforme Laboratorio 8910-2josue riveraAún no hay calificaciones
- Diseño de primers para la amplificación de genes mediante programas bioinformáticosDocumento18 páginasDiseño de primers para la amplificación de genes mediante programas bioinformáticosVANESSA LÓPEZ LÓPEZAún no hay calificaciones
- Diseño de PrimerDocumento6 páginasDiseño de PrimerJessica Hernandez SanchezAún no hay calificaciones
- PCR FundamentoDocumento10 páginasPCR FundamentoEillenSáezAún no hay calificaciones
- Trabajo de BIOINFORMATICADocumento19 páginasTrabajo de BIOINFORMATICANathaly RomeroAún no hay calificaciones
- t7 Genética Molecular. El ADN Como Mensajero GenéticoDocumento9 páginast7 Genética Molecular. El ADN Como Mensajero GenéticoMarina RMAún no hay calificaciones
- Taller 1 F.FDocumento7 páginasTaller 1 F.FYair VillanuevaAún no hay calificaciones
- Guía de Problemas - 1ra Parte - BCM Nutricion 2023 - Clase 1Documento8 páginasGuía de Problemas - 1ra Parte - BCM Nutricion 2023 - Clase 1leonardo farioliAún no hay calificaciones
- 2reporte GeneticaDocumento5 páginas2reporte GeneticaDaniel MendozaAún no hay calificaciones
- Laboratorio de Ing. GenéticaDocumento28 páginasLaboratorio de Ing. GenéticaAntoniaAún no hay calificaciones
- PCR-RT: Amplificación de ARNDocumento9 páginasPCR-RT: Amplificación de ARNvioletaAún no hay calificaciones
- Taller Replicación-Transcripción-TraducciónDocumento6 páginasTaller Replicación-Transcripción-TraducciónSantiago OssaAún no hay calificaciones
- Práctica Xiii XVDocumento16 páginasPráctica Xiii XVJUBER CALEB TOVAR VARGASAún no hay calificaciones
- 4to Informe - Grupo 2Documento16 páginas4to Informe - Grupo 2Emmanuel Oscar ALBINES SECLENAún no hay calificaciones
- Trabajo de Biologia II - RAPDDocumento11 páginasTrabajo de Biologia II - RAPDmadrosal377Aún no hay calificaciones
- Obtención de secuencias y diseño de cebadoresDocumento9 páginasObtención de secuencias y diseño de cebadoresmariaAún no hay calificaciones
- Cuantificación Absoluta Del Transcriptoma de La Levadura - RESUMEN-INTRODUCCIÓNDocumento4 páginasCuantificación Absoluta Del Transcriptoma de La Levadura - RESUMEN-INTRODUCCIÓNJuan Yujra CárdenasAún no hay calificaciones
- P13 - Adn in VitroDocumento15 páginasP13 - Adn in Vitroalba.politecnico34Aún no hay calificaciones
- Act. 1.trabajo ColaborativoDocumento8 páginasAct. 1.trabajo ColaborativoAnonymous IG3Pj6AwAún no hay calificaciones
- Trabajo Final PCR y Secuenciación - TiradoZazuetaGuadalupeDocumento10 páginasTrabajo Final PCR y Secuenciación - TiradoZazuetaGuadalupeGuadalupe TZAún no hay calificaciones
- Resumen 4 Molecular BorradorDocumento3 páginasResumen 4 Molecular BorradorGio CañizaresAún no hay calificaciones
- GENETICA Seminario 3 BQIIDocumento7 páginasGENETICA Seminario 3 BQIILucia RamirezAún no hay calificaciones
- PCR - Reaccion en Cadena de La Polimerasa y AluDocumento9 páginasPCR - Reaccion en Cadena de La Polimerasa y AluArantxa González IturraAún no hay calificaciones
- Flujo de Información GenéticaDocumento2 páginasFlujo de Información GenéticaYaiza BetanyaAún no hay calificaciones
- Laboratorio Biología Celular Y Molecular: Análisis Molecular Por PCRDocumento5 páginasLaboratorio Biología Celular Y Molecular: Análisis Molecular Por PCRFLORENCIA ANTONIA AVALOS CONEJEROSAún no hay calificaciones
- Reacción en Cadena de La PolimerasaDocumento14 páginasReacción en Cadena de La PolimerasaAlejandro PazAún no hay calificaciones
- TALLER DE REACCIÓN EN CADENA DE LA POLIMERASA (PCR) (1) (2)Documento5 páginasTALLER DE REACCIÓN EN CADENA DE LA POLIMERASA (PCR) (1) (2)Rosa Isabel Guette TorneAún no hay calificaciones
- Amplificación masiva ADN mediante PCRDocumento3 páginasAmplificación masiva ADN mediante PCRDaniela Maria Barrientos GuzmanAún no hay calificaciones
- Tarea Sobre Traducción y MutacionesDocumento3 páginasTarea Sobre Traducción y MutacionesJANETH AURORA CRUZ VILLEGAS0% (1)
- Ciencias Taller #1Documento5 páginasCiencias Taller #1Juan Andres Quintero ClaroAún no hay calificaciones
- Mutaciones y splicing alternativo en la replicación del ADNDocumento8 páginasMutaciones y splicing alternativo en la replicación del ADNDhanur GaliscoAún no hay calificaciones
- Taller de Trascripcion y TraduccionDocumento7 páginasTaller de Trascripcion y TraduccionmcAún no hay calificaciones
- Laboratorio #3Documento9 páginasLaboratorio #3Sofia GarciaAún no hay calificaciones
- Taller 12 Acidos Nucleicos y Sintesis de ProteinasDocumento6 páginasTaller 12 Acidos Nucleicos y Sintesis de Proteinasalexander silvaAún no hay calificaciones
- Cuestiones de Aplicación PCRDocumento8 páginasCuestiones de Aplicación PCRLara Vázquez FafiánAún no hay calificaciones
- Herramientas de La Ingeniería GenéticaDocumento19 páginasHerramientas de La Ingeniería GenéticaEDILMAHERNANDEZAún no hay calificaciones
- Ingeniería Genética: Introducción y técnicasDocumento9 páginasIngeniería Genética: Introducción y técnicasluisetecampeonAún no hay calificaciones
- Informe - Informe 3Documento9 páginasInforme - Informe 3Diego Cruzado oroscoAún no hay calificaciones
- Secuenciación de ADN: historia y métodosDocumento22 páginasSecuenciación de ADN: historia y métodosDiego A. Benites PasaperaAún no hay calificaciones
- 5 Informe de Laboratorio - PCR y ElectroforesisDocumento11 páginas5 Informe de Laboratorio - PCR y ElectroforesisAndrews DiestraAún no hay calificaciones
- Trabajo Práctico: Reacción en Cadena de La Polimerasa B101Documento12 páginasTrabajo Práctico: Reacción en Cadena de La Polimerasa B101Ro LiliAún no hay calificaciones
- Taller TraduccionDocumento3 páginasTaller Traduccionnatalia andreaAún no hay calificaciones
- Biologia MolecularDocumento11 páginasBiologia MolecularAleximon GraoAún no hay calificaciones
- Técnicas Biología MolecularDocumento4 páginasTécnicas Biología MolecularJose Antonio Carballo Junco100% (1)
- Taller 9. Diseño de Primers 2021Documento14 páginasTaller 9. Diseño de Primers 2021CarlosAún no hay calificaciones
- TALLER 04 Genetica Molecular 2017Documento2 páginasTALLER 04 Genetica Molecular 2017Taydith Teresa Sierra Tibamoza0% (1)
- Clonación por PCR santiDocumento10 páginasClonación por PCR santibeod2115Aún no hay calificaciones
- Taller 4_2410 (1)Documento4 páginasTaller 4_2410 (1)Paula Sophía CastiblancoAún no hay calificaciones
- Reacción en Cadena de La Polimerasa y Diseño de IniciadoresDocumento44 páginasReacción en Cadena de La Polimerasa y Diseño de IniciadoresValeria Gutierrez RuizAún no hay calificaciones
- Adn y ArnDocumento12 páginasAdn y Arnefrain juliaoAún no hay calificaciones
- PCR DiapositivasDocumento15 páginasPCR DiapositivasJey Carbonell SanJuan100% (2)
- Practicas Clonación de Un GenDocumento5 páginasPracticas Clonación de Un GenDaryl TinjacaAún no hay calificaciones
- Informe Adn RecombinanteDocumento6 páginasInforme Adn Recombinantealejandra galeano100% (1)
- PCRDocumento6 páginasPCRJuliana CorderoAún no hay calificaciones
- Actividades de BM Unidad 2Documento2 páginasActividades de BM Unidad 2paulamontes2305Aún no hay calificaciones
- Genética - Segundo Parcial SumativaDocumento5 páginasGenética - Segundo Parcial SumativaSergio MedellinAún no hay calificaciones
- Código genético y mutacionesDocumento3 páginasCódigo genético y mutacionesItala RoncoAún no hay calificaciones
- Introducción a la Biología: RESÚMENES UNIVERSITARIOSDe EverandIntroducción a la Biología: RESÚMENES UNIVERSITARIOSCalificación: 5 de 5 estrellas5/5 (1)
- Neurotransmisores y neuromoduladores: tipos y mecanismosDocumento23 páginasNeurotransmisores y neuromoduladores: tipos y mecanismosKaren EstevesAún no hay calificaciones
- Formando personas con valores y habilidades para contribuir a la sociedadDocumento41 páginasFormando personas con valores y habilidades para contribuir a la sociedadKaren EstevesAún no hay calificaciones
- Diversidad de Angiospermas: Magnoliophytas y MonocotiledoneasDocumento57 páginasDiversidad de Angiospermas: Magnoliophytas y MonocotiledoneasKaren EstevesAún no hay calificaciones
- Sistema CardiovascularDocumento32 páginasSistema CardiovascularKaren EstevesAún no hay calificaciones
- Clase 10 Evolución Plantas VascularesDocumento33 páginasClase 10 Evolución Plantas VascularesKaren EstevesAún no hay calificaciones
- Mecanismo de AcciónDocumento2 páginasMecanismo de AcciónKaren EstevesAún no hay calificaciones
- Sello formativo y misión institucionalDocumento3 páginasSello formativo y misión institucionalKaren EstevesAún no hay calificaciones
- Fases para La Estrategia de BúsquedaDocumento5 páginasFases para La Estrategia de BúsquedaKaren EstevesAún no hay calificaciones
- Clase 12 Diversidad de Plantas Leñosas y GimnospermasDocumento34 páginasClase 12 Diversidad de Plantas Leñosas y GimnospermasKaren EstevesAún no hay calificaciones
- CONTROLDocumento20 páginasCONTROLKaren EstevesAún no hay calificaciones
- INTRODUCCIONDocumento6 páginasINTRODUCCIONKaren EstevesAún no hay calificaciones
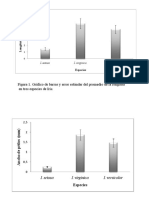
- Figura 1. Gráfico de Barras y Error Estándar Del Promedio de La Longitud en Tres Especies de IrisDocumento4 páginasFigura 1. Gráfico de Barras y Error Estándar Del Promedio de La Longitud en Tres Especies de IrisKaren EstevesAún no hay calificaciones
- Comunicacion No VerbalDocumento38 páginasComunicacion No VerbalKaren EstevesAún no hay calificaciones
- Autogestión - Carolina OsorioDocumento3 páginasAutogestión - Carolina OsorioKaren EstevesAún no hay calificaciones
- Economía para No Economistas - CompletoDocumento353 páginasEconomía para No Economistas - CompletoChipi Busson100% (5)
- 04 EC-DIAZ Capitulo1Documento16 páginas04 EC-DIAZ Capitulo1Florencia PalombaAún no hay calificaciones
- Lesp06 U1 GaDocumento29 páginasLesp06 U1 GamaquensyAún no hay calificaciones
- Concepto Básicos de PresupuestoDocumento32 páginasConcepto Básicos de PresupuestoKaren EstevesAún no hay calificaciones
- Clase 1Documento28 páginasClase 1Karen EstevesAún no hay calificaciones
- Empresa y ContabilidadDocumento57 páginasEmpresa y ContabilidadKaren EstevesAún no hay calificaciones
- Hacer lo legal es éticoDocumento6 páginasHacer lo legal es éticoKaren EstevesAún no hay calificaciones
- Resuelta COMPUDocumento2 páginasResuelta COMPUKaren EstevesAún no hay calificaciones
- Política Económica - Ley de Protección Al EmpleoDocumento4 páginasPolítica Económica - Ley de Protección Al EmpleoKaren EstevesAún no hay calificaciones
- Paquete CancúnDocumento3 páginasPaquete CancúnKaren EstevesAún no hay calificaciones
- Apunte EstadisticaDocumento22 páginasApunte EstadisticaKaren EstevesAún no hay calificaciones
- Control 1 Ifrs - Karen Esteves - Sección 30Documento3 páginasControl 1 Ifrs - Karen Esteves - Sección 30Karen EstevesAún no hay calificaciones
- Clase 1Documento28 páginasClase 1Karen EstevesAún no hay calificaciones
- Semana 1 - Tecnología de La InformaciónDocumento12 páginasSemana 1 - Tecnología de La InformaciónKaren EstevesAún no hay calificaciones
- Semana 1 - Teclado ExpandidoDocumento10 páginasSemana 1 - Teclado ExpandidoKaren EstevesAún no hay calificaciones
- Presentación 2 AutogestionDocumento27 páginasPresentación 2 AutogestionKaren EstevesAún no hay calificaciones
- El Camino A La Doble Hélice - CurtisDocumento3 páginasEl Camino A La Doble Hélice - CurtisOsoPandoAún no hay calificaciones
- Fish WordDocumento3 páginasFish WordCarolina ElguetaAún no hay calificaciones
- Adn, Arn Mitosis y MeiosisDocumento20 páginasAdn, Arn Mitosis y MeiosisAndres ParedesAún no hay calificaciones
- Farmacocinetica y FarmacodinamiaDocumento41 páginasFarmacocinetica y Farmacodinamiaraiza torresAún no hay calificaciones
- Diapositivas CorreguidasDocumento18 páginasDiapositivas CorreguidasCardonaCristianAún no hay calificaciones
- Laboratorio de MitosisDocumento13 páginasLaboratorio de MitosisVALENTINA CASTILLO GUERREROAún no hay calificaciones
- Avance y Perspectivas de La Biotecnología en Yucatán 2018Documento542 páginasAvance y Perspectivas de La Biotecnología en Yucatán 2018Enrique SauriAún no hay calificaciones
- Avances en el diagnóstico de laboratorio de virusDocumento3 páginasAvances en el diagnóstico de laboratorio de virusANDREA MOYA MACIELAún no hay calificaciones
- Examen Trimestral Del Curso de BiologiaDocumento2 páginasExamen Trimestral Del Curso de BiologiaGustavo Huaman VilchezAún no hay calificaciones
- Cultivoscelularesanticuerposmonoclonares 130803210924 Phpapp02Documento18 páginasCultivoscelularesanticuerposmonoclonares 130803210924 Phpapp02Abelardo Möller100% (1)
- Sintesis de Acidos GrasosDocumento12 páginasSintesis de Acidos GrasosKARLA VERONICA DIAZ AMAYAAún no hay calificaciones
- PREICFES 9 Ciencias NaturalesDocumento3 páginasPREICFES 9 Ciencias NaturalesEstefania SalazarAún no hay calificaciones
- Genómica funcional: estudio dinámico de genes y sus funcionesDocumento9 páginasGenómica funcional: estudio dinámico de genes y sus funcionesFrancisco DiazAún no hay calificaciones
- La Célula 1. Explique La Teoría Endosimbióntica en Serie de Lynn MargulisDocumento3 páginasLa Célula 1. Explique La Teoría Endosimbióntica en Serie de Lynn MargulisJuanJoMurilloAún no hay calificaciones
- 1 ProteinasDocumento10 páginas1 ProteinasMartha Lia Castaño EcheverryAún no hay calificaciones
- La Membrana Celular y La Matriz Extracelular JorgeDocumento8 páginasLa Membrana Celular y La Matriz Extracelular JorgeJenifer AcostaAún no hay calificaciones
- Impactos Ambientales Generados en Plantas de Beneficio BovinoDocumento65 páginasImpactos Ambientales Generados en Plantas de Beneficio BovinoRuther Miranda MAún no hay calificaciones
- Cloroplastos y Mitocondrias Quimica 2019-1Documento37 páginasCloroplastos y Mitocondrias Quimica 2019-1Adan Vega Alvarez0% (1)
- Suero Fetal BovinoDocumento10 páginasSuero Fetal BovinococoAún no hay calificaciones
- CromatinaDocumento15 páginasCromatinaDulce García DuránAún no hay calificaciones
- Reporte de Vacunas - RED DE SALUD TARMADocumento14 páginasReporte de Vacunas - RED DE SALUD TARMAmanuel jesus rivera fernandezAún no hay calificaciones
- Biologia Celular Resumen.Documento42 páginasBiologia Celular Resumen.Nie LiAún no hay calificaciones
- Replicacion - Transcripcion, Traduccion y Codigo GeneticoDocumento38 páginasReplicacion - Transcripcion, Traduccion y Codigo GeneticoDooominic0% (1)
- Comisión de Apuntes Biología Celular, Histología y Organografía.Documento164 páginasComisión de Apuntes Biología Celular, Histología y Organografía.Melissa VelásquezAún no hay calificaciones
- Glucólisis - Fraccionamiento - Grupo N°3Documento19 páginasGlucólisis - Fraccionamiento - Grupo N°3Andres carpioAún no hay calificaciones
- Cromosoma: estructura y funciónDocumento3 páginasCromosoma: estructura y funciónCarlaAún no hay calificaciones
- A. Ecología Microbiana A 2018Documento119 páginasA. Ecología Microbiana A 2018Sebastian R. ToroAún no hay calificaciones
- Dogma CentralDocumento21 páginasDogma CentralBlanca VargasAún no hay calificaciones
- 2016-II Silabo Genetica IDocumento12 páginas2016-II Silabo Genetica Iesteban zapataAún no hay calificaciones