También podría gustarte
- Tumor CerebralDocumento4 páginasTumor CerebralHermes Shanel GeronimoAún no hay calificaciones
- Fisiopatología de La Diabetes Tipo 2Documento4 páginasFisiopatología de La Diabetes Tipo 2Luis Miguel Marlo EstelaAún no hay calificaciones
- Hemoptisis - Lucas RomanDocumento9 páginasHemoptisis - Lucas RomanLucas RománAún no hay calificaciones
- Inflamación y ReparaciónDocumento120 páginasInflamación y ReparaciónErick Antonio Castillo GurdianAún no hay calificaciones
- Jurisdicción Voluntaria Diligencias Adperpetuam Perdida de Factura de Un VehiculoDocumento2 páginasJurisdicción Voluntaria Diligencias Adperpetuam Perdida de Factura de Un VehiculoLuis Demetrio Novelo100% (1)
- Anaya Antonio Manuel Reyes Rodriguez CYPE 2010 Instalaciones Del Edificio y Cumplimiento Del CTE PDFDocumento418 páginasAnaya Antonio Manuel Reyes Rodriguez CYPE 2010 Instalaciones Del Edificio y Cumplimiento Del CTE PDFJoel Antonio Quezada MejiaAún no hay calificaciones
- Cuestionario Semana 4Documento4 páginasCuestionario Semana 4Joyas Mar Pau100% (2)
- Infecciones Respiratorias Altas (Resumen 1) - 1Documento10 páginasInfecciones Respiratorias Altas (Resumen 1) - 1Nathaly Laguna AlvaradoAún no hay calificaciones
- Apnea y AsfixiaDocumento11 páginasApnea y AsfixiaXóchitl Adriana Contreras GómezAún no hay calificaciones
- Tratamiento de Aguas ResidualesDocumento234 páginasTratamiento de Aguas ResidualesJosé Carlos Cardenas Palacios50% (2)
- Manejo de InsulinaDocumento5 páginasManejo de InsulinaPlinio MendozaAún no hay calificaciones
- Ejercicios en Mineria SuperficialDocumento5 páginasEjercicios en Mineria Superficialwilscor100% (1)
- Propedéutica RespiratoriaDocumento26 páginasPropedéutica RespiratoriaMycroft HolmesAún no hay calificaciones
- Clase 1 Fisiologia de La SangreDocumento109 páginasClase 1 Fisiologia de La SangreNia Avellaneda100% (1)
- Shock Hemorragico Articulo PDFDocumento22 páginasShock Hemorragico Articulo PDFEstefaniSabrinaSandovalAún no hay calificaciones
- Traducción en EucariontesDocumento3 páginasTraducción en EucariontesRenzo Cortez PachecoAún no hay calificaciones
- Alteraciones de La Función RespiratoriaDocumento50 páginasAlteraciones de La Función RespiratoriaJonathan Martinez MaldonadoAún no hay calificaciones
- Corazón PDF 1Documento145 páginasCorazón PDF 1Omar Leonardo Guerrero SuarezAún no hay calificaciones
- DETERMINACION DEL HEMATÓCRITO Y HEMOGLOBINA BioquimicaDocumento5 páginasDETERMINACION DEL HEMATÓCRITO Y HEMOGLOBINA BioquimicaAndres Camilo Moreno GarciaAún no hay calificaciones
- ICTERICIADocumento5 páginasICTERICIAlazernxAún no hay calificaciones
- Estudio de La Función Hepática. Magnitudes BioquímicasDocumento25 páginasEstudio de La Función Hepática. Magnitudes BioquímicasCarlosEnriqueVegaSanchezAún no hay calificaciones
- Monografia Fibrosis PumonarDocumento25 páginasMonografia Fibrosis PumonarRoger Juan Vera Gutierrez100% (1)
- FAGOCITOSISDocumento4 páginasFAGOCITOSISAngel Ramiro Soplin AlmeidaAún no hay calificaciones
- Resumen-Fisiología de La Secreción PancreáticaDocumento3 páginasResumen-Fisiología de La Secreción PancreáticaJose Alfredo HernandezAún no hay calificaciones
- B. - Eosinofilia y Parasitismo-1Documento8 páginasB. - Eosinofilia y Parasitismo-1Brian AcostaAún no hay calificaciones
- Fisiopatología-Insuficiencia HepaticaDocumento4 páginasFisiopatología-Insuficiencia HepaticaJuan Sebastian Montoya FernándezAún no hay calificaciones
- Tema 9 Fisiopatologia de Las Vias Biliares y Pancreas ExocrinoDocumento7 páginasTema 9 Fisiopatologia de Las Vias Biliares y Pancreas Exocrinoana paula fernandez acostaAún no hay calificaciones
- Anatomia PDFDocumento132 páginasAnatomia PDFJoffre Jeorwin Cartuche CalvaAún no hay calificaciones
- Reg. Prof. Meninges, Senos Durales, Cavidades IntraencefalicasDocumento11 páginasReg. Prof. Meninges, Senos Durales, Cavidades Intraencefalicasbetic094Aún no hay calificaciones
- Tos, Disnea, Cianosis, Atelectasia Fisiopatología Respiratoria. DR CasanovaDocumento82 páginasTos, Disnea, Cianosis, Atelectasia Fisiopatología Respiratoria. DR CasanovaRenzo Donato Sairitupa RamirezAún no hay calificaciones
- Trabajo Integrador Final - Sistema RespiratorioDocumento16 páginasTrabajo Integrador Final - Sistema RespiratorioGabriel Carp PalazzoloAún no hay calificaciones
- Presentacion HepatitisDocumento34 páginasPresentacion HepatitisLenin AvilésAún no hay calificaciones
- Diabetes Mullitus PDFDocumento66 páginasDiabetes Mullitus PDFFelix Manuel Romero PalaciosAún no hay calificaciones
- Liquidos y Electrolitos UTESADocumento68 páginasLiquidos y Electrolitos UTESAEdgar LunaAún no hay calificaciones
- Articulo Sobre Profilaxis QuirurgicaDocumento7 páginasArticulo Sobre Profilaxis QuirurgicaRoberto FiallosAún no hay calificaciones
- Causas HiperuricemiaDocumento2 páginasCausas HiperuricemiaMaximiliam RobespierreAún no hay calificaciones
- Nutiricion EnteralDocumento32 páginasNutiricion EnteralMaria LauraAún no hay calificaciones
- Rombo de SeguridadDocumento9 páginasRombo de Seguridadquieroleer888Aún no hay calificaciones
- Adenovirus JACL Microypara 2011ADocumento25 páginasAdenovirus JACL Microypara 2011AJocelyn CabralesAún no hay calificaciones
- Pruebas Funcionales RespiratoriasDocumento6 páginasPruebas Funcionales RespiratoriasDiego Ast HnAún no hay calificaciones
- Qué Pasa Con El Cuerpo Cuando Hacemos EjercicioDocumento7 páginasQué Pasa Con El Cuerpo Cuando Hacemos EjercicioYesi BenitezAún no hay calificaciones
- HIPERTENSIÓN PORTAL FinalDocumento20 páginasHIPERTENSIÓN PORTAL FinalJeaneth AniAún no hay calificaciones
- Hipertensión Portal para SeminarioDocumento70 páginasHipertensión Portal para SeminarioCiro Calviño EstradaAún no hay calificaciones
- Enfisema PulmonarDocumento40 páginasEnfisema PulmonarAremy Esther CortésAún no hay calificaciones
- Historia Clinica SemiologiaDocumento49 páginasHistoria Clinica SemiologiaJean Legrand100% (1)
- Hematología 1Documento29 páginasHematología 1kaleyviAún no hay calificaciones
- TrombosisDocumento12 páginasTrombosisYamilet CaamañoAún no hay calificaciones
- CesarDocumento10 páginasCesarapi-3699557100% (1)
- Respuesta Adquirida (Inmunidad Adaptativa)Documento17 páginasRespuesta Adquirida (Inmunidad Adaptativa)Orbely Garcia100% (1)
- SX de Absorción Intestinal Deficiente.Documento34 páginasSX de Absorción Intestinal Deficiente.Ariadna MarielAún no hay calificaciones
- Drenaje ToracicoDocumento7 páginasDrenaje Toracicopaulino100% (1)
- Fisiologia de La Hemostasia y Coagulacion SanguineaDocumento6 páginasFisiologia de La Hemostasia y Coagulacion SanguineaYong MondragonAún no hay calificaciones
- Sinapsis NeuromuscularDocumento4 páginasSinapsis NeuromuscularFabio SotoAún no hay calificaciones
- Absorción de Fármacos PDFDocumento45 páginasAbsorción de Fármacos PDFJulieta MoraAún no hay calificaciones
- Lupus Eritematoso SistemáticoDocumento12 páginasLupus Eritematoso SistemáticoLuisa Fernanda Campos Briceño100% (1)
- Mediastino PDFDocumento11 páginasMediastino PDFCalmantesMontesAún no hay calificaciones
- Tabla de ParásitosDocumento12 páginasTabla de Parásitosyose100% (1)
- Virus Lentos No ConvencionalesDocumento11 páginasVirus Lentos No Convencionalesmiguel110198250% (2)
- FagocitosisDocumento10 páginasFagocitosispukka38Aún no hay calificaciones
- Pta 2-11-2021Documento14 páginasPta 2-11-2021JOSE DANILO TABARQUINO LADINOAún no hay calificaciones
- LeucemiaDocumento40 páginasLeucemiaChac OregonAún no hay calificaciones
- Gestión Del Cuidado en Paciente Drenaje Pleural 2Documento37 páginasGestión Del Cuidado en Paciente Drenaje Pleural 2Melissa Rodriguez VillafanAún no hay calificaciones
- Tratamiento Farmacológico Frente Al PaludismoDocumento30 páginasTratamiento Farmacológico Frente Al PaludismoDaisyAún no hay calificaciones
- Síndrome MetabólicoDocumento6 páginasSíndrome MetabólicoLaura Valentina GarcíaAún no hay calificaciones
- Piel, Heridas, Suturas y AnalgésicosDocumento62 páginasPiel, Heridas, Suturas y AnalgésicosAlexis GuerreroGuerreroAún no hay calificaciones
- EndometriosisDocumento25 páginasEndometriosisAlexis GuerreroGuerreroAún no hay calificaciones
- Insulina EndoDocumento36 páginasInsulina EndoAlexis GuerreroGuerreroAún no hay calificaciones
- Ictericia ObstructivaDocumento50 páginasIctericia ObstructivaAlexis GuerreroGuerreroAún no hay calificaciones
- Trisomia 2113 y 18Documento3 páginasTrisomia 2113 y 18Alexis GuerreroGuerreroAún no hay calificaciones
- Fibrosis QuìsticaDocumento2 páginasFibrosis QuìsticaAlexis GuerreroGuerreroAún no hay calificaciones
- HC Cirugía - Brenda AyalaDocumento13 páginasHC Cirugía - Brenda AyalaAlexis GuerreroGuerreroAún no hay calificaciones
- FlowchartDocumento1 páginaFlowchartAlexis GuerreroGuerreroAún no hay calificaciones
- RCP Infografia 283111 Downloable 839011Documento3 páginasRCP Infografia 283111 Downloable 839011Alexis GuerreroGuerreroAún no hay calificaciones
- Vih EmbarazoDocumento27 páginasVih EmbarazoAlexis GuerreroGuerreroAún no hay calificaciones
- Anato y Fisio RespiDocumento17 páginasAnato y Fisio RespiAlexis GuerreroGuerreroAún no hay calificaciones
- Sufrimiento Fetal 2Documento25 páginasSufrimiento Fetal 2Alexis GuerreroGuerreroAún no hay calificaciones
- Hipotiroidismo y NTDocumento19 páginasHipotiroidismo y NTAlexis GuerreroGuerreroAún no hay calificaciones
- Sufrimiento Fetal 2Documento25 páginasSufrimiento Fetal 2Alexis GuerreroGuerreroAún no hay calificaciones
- Registro TopocardiograficoDocumento15 páginasRegistro TopocardiograficoAlexis GuerreroGuerreroAún no hay calificaciones
- Sufrimiento Fetal 2Documento25 páginasSufrimiento Fetal 2Alexis GuerreroGuerreroAún no hay calificaciones
- Endometriosis 1Documento19 páginasEndometriosis 1Alexis GuerreroGuerreroAún no hay calificaciones
- Expo LinfoDocumento68 páginasExpo LinfoAlexis GuerreroGuerreroAún no hay calificaciones
- Cuadro Comparatvo RespiDocumento3 páginasCuadro Comparatvo RespiAlexis GuerreroGuerreroAún no hay calificaciones
- Registro TopocardiograficoDocumento15 páginasRegistro TopocardiograficoAlexis GuerreroGuerreroAún no hay calificaciones
- Embarazo en La AdolescenciaDocumento10 páginasEmbarazo en La AdolescenciaAlexis GuerreroGuerreroAún no hay calificaciones
- Ruptura Prematura de Membranas Expo BienDocumento36 páginasRuptura Prematura de Membranas Expo BienAlexis GuerreroGuerreroAún no hay calificaciones
- Fia Emenino FisiologDocumento8 páginasFia Emenino FisiologAlexis GuerreroGuerreroAún no hay calificaciones
- Registro TopocardiograficoDocumento15 páginasRegistro TopocardiograficoAlexis GuerreroGuerreroAún no hay calificaciones
- M. Practica Clinica IIIDocumento29 páginasM. Practica Clinica IIIAlexis GuerreroGuerreroAún no hay calificaciones
- Sufrimiento Fetal 2Documento25 páginasSufrimiento Fetal 2Alexis GuerreroGuerreroAún no hay calificaciones
- PotencialdeaccionDocumento20 páginasPotencialdeaccionAlexis GuerreroGuerreroAún no hay calificaciones
- Tráquea y Bronquios ExtrapulmonaresDocumento76 páginasTráquea y Bronquios ExtrapulmonaresAlexis GuerreroGuerreroAún no hay calificaciones
- Sistema Vascular PerifericoDocumento130 páginasSistema Vascular PerifericoAlexis GuerreroGuerreroAún no hay calificaciones
- Clase 3 AnticuerposDocumento41 páginasClase 3 AnticuerposAlexis GuerreroGuerreroAún no hay calificaciones
- Migración y Mercado Laboral en Chile (FEN UCH)Documento26 páginasMigración y Mercado Laboral en Chile (FEN UCH)AMpoltiosAún no hay calificaciones
- Antecedentes de La InvestigacionDocumento4 páginasAntecedentes de La InvestigacionMi estilo onlineAún no hay calificaciones
- Arbóreas y ArbustivasDocumento1 páginaArbóreas y ArbustivasYessenia Lagos RAún no hay calificaciones
- Stripping RatioDocumento42 páginasStripping RatioRicardo Querebalú Nevado100% (1)
- Piar 2023 Alejandro Carmona T.Documento3 páginasPiar 2023 Alejandro Carmona T.Edwins Chávez SamientoAún no hay calificaciones
- Formato Incripción Creacion de Empresa Version 2. 06.06.2018Documento6 páginasFormato Incripción Creacion de Empresa Version 2. 06.06.2018Paola Betancourt RubioAún no hay calificaciones
- Formulario de Postulacion FFOIP 2023Documento13 páginasFormulario de Postulacion FFOIP 2023Juan Ignacio MartínezAún no hay calificaciones
- COMPROBANTEDocumento1 páginaCOMPROBANTEJhonatan Aguilar VásquezAún no hay calificaciones
- Producto Académico #1 Gestión de OperacionesDocumento7 páginasProducto Académico #1 Gestión de OperacionesLuis Antonio Bravo SaucedoAún no hay calificaciones
- Equipo Panasonic® Con Error F61Documento17 páginasEquipo Panasonic® Con Error F61khalet100% (1)
- Clase 4.0 - Planificación Financiera PDFDocumento38 páginasClase 4.0 - Planificación Financiera PDFmauricioAún no hay calificaciones
- Nom 001 Cre Scfi 2019Documento214 páginasNom 001 Cre Scfi 2019David Moreno100% (1)
- Peña Marleny Mapa ConceptualDocumento1 páginaPeña Marleny Mapa ConceptualMarleny SantosAún no hay calificaciones
- ISO 9001 - 2015 Seccion ApoyoDocumento20 páginasISO 9001 - 2015 Seccion ApoyoJuankSalazarAún no hay calificaciones
- Ficha Tecnica Ultrasol SopDocumento1 páginaFicha Tecnica Ultrasol SopValencia HugoAún no hay calificaciones
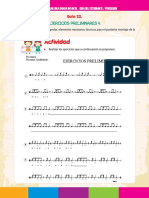
- Guia Estudiante Percusión PDFDocumento2 páginasGuia Estudiante Percusión PDFNatalia Garcia MorenoAún no hay calificaciones
- Manual Robot AspiradoraDocumento20 páginasManual Robot AspiradoraCarlosAún no hay calificaciones
- Zeolita Acqua Tecnologia PDFDocumento1 páginaZeolita Acqua Tecnologia PDFLeevan BauAún no hay calificaciones
- Informe Ampliación de Plazo INSPECCIÓNDocumento9 páginasInforme Ampliación de Plazo INSPECCIÓNLuis VelasqueAún no hay calificaciones
- Ejercicios Propuestos de DerivadasDocumento6 páginasEjercicios Propuestos de DerivadasMikeAún no hay calificaciones
- Cuestionario Capitulo Columnas CortasDocumento2 páginasCuestionario Capitulo Columnas Cortasrmuñoz_895671Aún no hay calificaciones
- Informe Mantención Compresor de TornillosDocumento10 páginasInforme Mantención Compresor de TornillosChristian Alfaro SanchezAún no hay calificaciones
- Funcion Si Winner GroupDocumento81 páginasFuncion Si Winner Groupandres cAún no hay calificaciones
- Unidad 2 - Tarea 2 - Dinámica y EnergíaDocumento8 páginasUnidad 2 - Tarea 2 - Dinámica y EnergíaDario BuitragoAún no hay calificaciones