También podría gustarte
- Farmacocinética LADME FARM 288Documento85 páginasFarmacocinética LADME FARM 288Valentina Cea LobosAún no hay calificaciones
- Dis TribDocumento23 páginasDis TribMicheellee Goomithaa Wii WiiAún no hay calificaciones
- 2.farmacocinética II MRCDocumento46 páginas2.farmacocinética II MRCjaviera riquelmeAún no hay calificaciones
- Farmacocinética y FarmacodinamiaDocumento54 páginasFarmacocinética y FarmacodinamiaJocelyn AlejandraAún no hay calificaciones
- Farmacocinetica GeneralDocumento3 páginasFarmacocinetica Generaltq9prx5s5qAún no hay calificaciones
- ADME Distribucion Metabolismo y ExcrecionDocumento6 páginasADME Distribucion Metabolismo y ExcrecionCristian Andrés VeraAún no hay calificaciones
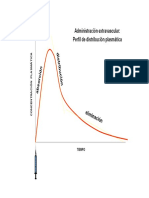
- Administración Extravascular: Administración Extravascular: Perfil de Distribución PlasmáticaDocumento36 páginasAdministración Extravascular: Administración Extravascular: Perfil de Distribución PlasmáticaNana SuárezAún no hay calificaciones
- Farmacocinetica y FarmacodinamiaDocumento37 páginasFarmacocinetica y FarmacodinamiaZulma Madeline L. Gutarra TicaAún no hay calificaciones
- Farmacocinética DrSalasDocumento98 páginasFarmacocinética DrSalasCesar Del Aguila MattaAún no hay calificaciones
- Distribución de FármacosDocumento61 páginasDistribución de FármacosJocelyn SalinasAún no hay calificaciones
- Resumen 1ra Prueba Farmacología y SNDocumento19 páginasResumen 1ra Prueba Farmacología y SNjuan carlosAún no hay calificaciones
- FARMACOCINETICA y FARMACODINAMIADocumento25 páginasFARMACOCINETICA y FARMACODINAMIANadia MoscaAún no hay calificaciones
- FarmacocinéticaDocumento54 páginasFarmacocinéticaJean Pierre Cano MolinaAún no hay calificaciones
- FARMACOLOGIA Y TERAPEUTICA VETERINARIA I.ppt 11Documento197 páginasFARMACOLOGIA Y TERAPEUTICA VETERINARIA I.ppt 11Adolfo Canaza T100% (1)
- Sistema Ladme Expo.Documento16 páginasSistema Ladme Expo.Ventocilla Castillo Itachi33% (3)
- Clases de FarmacologiaDocumento51 páginasClases de FarmacologiaKevin Brian LVAún no hay calificaciones
- 2 PARCIAL Fundamentos 3 (Recuperado Automáticamente)Documento15 páginas2 PARCIAL Fundamentos 3 (Recuperado Automáticamente)Keren ParejoAún no hay calificaciones
- FarmacocineticaDocumento61 páginasFarmacocineticaalvaro alejandro restrepo zapataAún no hay calificaciones
- FARMACOCINÉTICA Part2Documento6 páginasFARMACOCINÉTICA Part2Danny DavidAún no hay calificaciones
- 04 Metabolismo de FarmacosDocumento48 páginas04 Metabolismo de FarmacosTaller De Tesis Farmacia UladechAún no hay calificaciones
- Farmacocinética IIDocumento10 páginasFarmacocinética IIEFRAIN ANTONIO CASTAÑEDA MACIASAún no hay calificaciones
- Clase 5. Farmacocinética - Absorción y DistribuciónDocumento30 páginasClase 5. Farmacocinética - Absorción y DistribuciónVanessa ortiz GuevaraAún no hay calificaciones
- FarmacocineticaDocumento23 páginasFarmacocineticaMargot Mendoza SalasAún no hay calificaciones
- CLASE 2 - Farmacocinética II y Farmacodinamia - 2022Documento43 páginasCLASE 2 - Farmacocinética II y Farmacodinamia - 2022Danitza Paula100% (1)
- Transportadores de Membrana y Respuesta A FármacosDocumento84 páginasTransportadores de Membrana y Respuesta A FármacosEduardo Ortiz100% (3)
- Farmacocinética de Medicamentos 2022Documento76 páginasFarmacocinética de Medicamentos 2022catalina palaciosAún no hay calificaciones
- Clase 05. 26-10-18. Distribucion de Los FarmacosDocumento7 páginasClase 05. 26-10-18. Distribucion de Los FarmacoslorenaAún no hay calificaciones
- Taller 2 FarmacocinéticaDocumento39 páginasTaller 2 FarmacocinéticaLiz MedinaAún no hay calificaciones
- Modelos FarmacocinéticosDocumento18 páginasModelos Farmacocinéticosjbarreto75Aún no hay calificaciones
- Farmacocinetica de La Cruz Urgell Aldo JDocumento8 páginasFarmacocinetica de La Cruz Urgell Aldo JUrgellAún no hay calificaciones
- AnatoDocumento5 páginasAnatocarmenAún no hay calificaciones
- Farmacocinética FP ActualizadoDocumento47 páginasFarmacocinética FP ActualizadoheleanyAún no hay calificaciones
- Absorción, Distribución, Metabolismo y ExcreciónDocumento39 páginasAbsorción, Distribución, Metabolismo y ExcreciónAndrea Blanco EstradaAún no hay calificaciones
- Taller de FarmacocinéticaDocumento10 páginasTaller de Farmacocinética0scarkAún no hay calificaciones
- Sesión 1 y 2 - Farmacocinética I y IIDocumento95 páginasSesión 1 y 2 - Farmacocinética I y IIJulio Daniel Pacheco AlcántaraAún no hay calificaciones
- Tarea 3 - FARMACOCINETICA 2Documento8 páginasTarea 3 - FARMACOCINETICA 2Mario SegoviaAún no hay calificaciones
- Clase001 Introducción FarmacodinámiaDocumento66 páginasClase001 Introducción Farmacodinámiaapi-3710186Aún no hay calificaciones
- FARMACOCINETICADocumento23 páginasFARMACOCINETICAPARI MAYTA SARAH EVELYNAún no hay calificaciones
- Resumen Capitulo 3 de FarmacoDocumento20 páginasResumen Capitulo 3 de Farmacoamygalv08Aún no hay calificaciones
- Farmacooooooo 1.pptx )Documento52 páginasFarmacooooooo 1.pptx )Mario RuizAún no hay calificaciones
- PsicofarmacologíaDocumento66 páginasPsicofarmacologíafiel2003Aún no hay calificaciones
- Distribucion de Los Farmacos DefDocumento94 páginasDistribucion de Los Farmacos Defxime_benavides6116Aún no hay calificaciones
- Farmacología Aplicada A La Enfermería - 2023 Unidad #3: Farmacocinética: Proceso ADME, Liberación Desde Las Formas Farmacéuticas, Ventajas yDocumento17 páginasFarmacología Aplicada A La Enfermería - 2023 Unidad #3: Farmacocinética: Proceso ADME, Liberación Desde Las Formas Farmacéuticas, Ventajas yLautaro GaleanoAún no hay calificaciones
- FARMACOCINETICADocumento52 páginasFARMACOCINETICAalberto nidomeAún no hay calificaciones
- Teoricos y Talleres 2023Documento1117 páginasTeoricos y Talleres 2023Aldana Jazmin FrancoAún no hay calificaciones
- Clase 3 - FarmacodinamiaDocumento43 páginasClase 3 - FarmacodinamiaRoberto F. GameiroAún no hay calificaciones
- FARMACOLOGIA - GeneralidadesDocumento47 páginasFARMACOLOGIA - GeneralidadesJulio Andre Pizarro GiraoAún no hay calificaciones
- Quimis.y - Distribución y Eliminacion de FarmacosDocumento19 páginasQuimis.y - Distribución y Eliminacion de FarmacosNatán MarcilloAún no hay calificaciones
- Biodisponibilidad y Volumen de DistribucionDocumento26 páginasBiodisponibilidad y Volumen de Distribucionantony1falconAún no hay calificaciones
- Taller de FarmacocineticaDocumento11 páginasTaller de FarmacocineticaEdith Ramírez menAún no hay calificaciones
- Fichas de FarmacologíaDocumento55 páginasFichas de FarmacologíaCris SanchezAún no hay calificaciones
- Distribucion de FarmacosDocumento39 páginasDistribucion de FarmacosiVANAún no hay calificaciones
- Absorción de MedicamentosDocumento26 páginasAbsorción de Medicamentosmiguelgaistardo348Aún no hay calificaciones
- Distribución de FármacosDocumento10 páginasDistribución de FármacosDrika WattpadAún no hay calificaciones
- DistribuciónDocumento59 páginasDistribuciónarma.bril16Aún no hay calificaciones
- 3 Farmacocinética Ii - FarmacodinamiaDocumento20 páginas3 Farmacocinética Ii - FarmacodinamiaYenny Delgado GomezAún no hay calificaciones
- Acceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017De EverandAcceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017Aún no hay calificaciones
- Rejuvenecer Con El Plasma Sanguíneo De Los JóvenesDe EverandRejuvenecer Con El Plasma Sanguíneo De Los JóvenesCalificación: 5 de 5 estrellas5/5 (1)
- Coagulación Para Todos: Medicina Para TodosDe EverandCoagulación Para Todos: Medicina Para TodosCalificación: 4.5 de 5 estrellas4.5/5 (3)
- Jahs Min 2012/12/9 Bootstrap f2 0:55:20: Analyst Time Unit Date Method TimeDocumento9 páginasJahs Min 2012/12/9 Bootstrap f2 0:55:20: Analyst Time Unit Date Method TimeSl SantyAún no hay calificaciones
- Bolus Intravenoso Modelo Monocompartimental Datos Plasmáticos Y UrinariosDocumento27 páginasBolus Intravenoso Modelo Monocompartimental Datos Plasmáticos Y UrinariosSl SantyAún no hay calificaciones
- Ejercicios ResueltosDocumento7 páginasEjercicios ResueltosSl SantyAún no hay calificaciones
- Orden Cero Orden Uno SinkDocumento3 páginasOrden Cero Orden Uno SinkSl SantyAún no hay calificaciones
- UntitledDocumento76 páginasUntitledSl SantyAún no hay calificaciones
- Clase 7.2 Ejemplo Ejercicio de InfusiónDocumento2 páginasClase 7.2 Ejemplo Ejercicio de InfusiónEsmeralda QuezadaAún no hay calificaciones
- Ejercicio Datos OrinaDocumento1 páginaEjercicio Datos OrinaSl SantyAún no hay calificaciones
- Clearance de ÓrganosDocumento3 páginasClearance de ÓrganosSl SantyAún no hay calificaciones
- Bioexenciones 2022Documento23 páginasBioexenciones 2022Sl SantyAún no hay calificaciones
- Administración Ev. Dosis Múltiple - Modelo Monocompartimental 2022Documento32 páginasAdministración Ev. Dosis Múltiple - Modelo Monocompartimental 2022Sl SantyAún no hay calificaciones
- Bioequivalencia IDocumento30 páginasBioequivalencia ISl SantyAún no hay calificaciones
- 133-Portada FC Vol4num4.InddDocumento6 páginas133-Portada FC Vol4num4.Inddthalia troncosAún no hay calificaciones
- Uso Racional de Antibioticos Dia Mundial de La Salud OPS 2011 ADocumento31 páginasUso Racional de Antibioticos Dia Mundial de La Salud OPS 2011 AMeyi Guanilo YalicoAún no hay calificaciones
- Ministerio de Salud Resolucion 4445 de 1996 para Establecimientos Pres Tad Ores de Servicio de SaludDocumento32 páginasMinisterio de Salud Resolucion 4445 de 1996 para Establecimientos Pres Tad Ores de Servicio de SaludnelsonsupelanoAún no hay calificaciones
- Vías de AdministraciónDocumento29 páginasVías de Administracióniaeste.ecuadorAún no hay calificaciones
- Cronograma 2024 - Farma Industrial - 1er3er CuatrDocumento2 páginasCronograma 2024 - Farma Industrial - 1er3er CuatrfarmferrerofedericoAún no hay calificaciones
- Tarea 4 BiofarmaciaDocumento12 páginasTarea 4 BiofarmaciaJosé LuisAún no hay calificaciones
- Categorías de Medicamentos en El EmbarazoDocumento16 páginasCategorías de Medicamentos en El EmbarazoAnali UriasAún no hay calificaciones
- UntitledDocumento5 páginasUntitledArturo IsuhuaylasAún no hay calificaciones
- Quiz Admon de MedicamentosDocumento1 páginaQuiz Admon de MedicamentosAndrea BarreraAún no hay calificaciones
- Offarm: EditorialDocumento1 páginaOffarm: Editorialyusmicarmen almarzaAún no hay calificaciones
- Protocolo de Charol de ParoDocumento19 páginasProtocolo de Charol de ParoantonioAún no hay calificaciones
- Historia de La Salud Publica en BoliviaDocumento8 páginasHistoria de La Salud Publica en BoliviaGERMANAún no hay calificaciones
- Software Control de de 5c2b4s ExcelDocumento24 páginasSoftware Control de de 5c2b4s ExcelFabricioHerediaLozaAún no hay calificaciones
- Alerta 38-21Documento1 páginaAlerta 38-21Susana Del Rocio RGAún no hay calificaciones
- Actividad 6 - Logros y Aspectos A Mejorar en El Abordaje Del Problema. Conclusiones.Documento9 páginasActividad 6 - Logros y Aspectos A Mejorar en El Abordaje Del Problema. Conclusiones.Vanessa Nava100% (2)
- Administracion de FarmacosDocumento26 páginasAdministracion de FarmacosJuanita Lopez GuevaraAún no hay calificaciones
- Uso Racional de MedicamentosDocumento37 páginasUso Racional de Medicamentoswilmer81280Aún no hay calificaciones
- Lista de Precios $$ - Sumisalud Al 13-09-2021 (REVISADO)Documento6 páginasLista de Precios $$ - Sumisalud Al 13-09-2021 (REVISADO)Laura PeñaAún no hay calificaciones
- Sistema Institucional de Información Del Servicio Farmacéutico. G3Documento5 páginasSistema Institucional de Información Del Servicio Farmacéutico. G3Steven Ðe J. Perez MenesesAún no hay calificaciones
- Farma CareDocumento21 páginasFarma CareDAPHNE RAMOS CORTESAún no hay calificaciones
- Central de ComprasDocumento26 páginasCentral de ComprasMarta GAún no hay calificaciones
- Sesion 6Documento20 páginasSesion 6YANELI YOSALIN MERCADO VALENCIAAún no hay calificaciones
- Elaboracion de Formas FarmaceuticasDocumento6 páginasElaboracion de Formas FarmaceuticasrgarayarfAún no hay calificaciones
- Ley Del Ejercicio de La OLey Del Ejercicio de La Odontologíadontología LEO. FORENSEDocumento10 páginasLey Del Ejercicio de La OLey Del Ejercicio de La Odontologíadontología LEO. FORENSEMarihanne RamirezAún no hay calificaciones
- Atención de Urgencias.1Documento10 páginasAtención de Urgencias.1Jason Gomez100% (2)
- Marco LegalDocumento26 páginasMarco LegallewisantonyAún no hay calificaciones
- Ud 1 Configuracion Formativa Del Sector SanitarioDocumento18 páginasUd 1 Configuracion Formativa Del Sector SanitariolourdesAún no hay calificaciones
- proyecto-VISTA-ALEGRE 2Documento29 páginasproyecto-VISTA-ALEGRE 2NoeliaAún no hay calificaciones
- Buenas Practicas Farmaindustria PDFDocumento84 páginasBuenas Practicas Farmaindustria PDFEva DiéguezAún no hay calificaciones
- Mapro Hospital PampasDocumento5 páginasMapro Hospital PampasMarcel José Madrid DávilaAún no hay calificaciones