También podría gustarte
- Prolongando La Vida Del Paciente Con DiabetesDe EverandProlongando La Vida Del Paciente Con DiabetesAún no hay calificaciones
- Tamizaje NeonatalDocumento5 páginasTamizaje NeonatalBrisila PesantezAún no hay calificaciones
- Endocrinopatías, Embarazo y AnestesiaDocumento45 páginasEndocrinopatías, Embarazo y AnestesiaLizzeth Inés Sánchez MendozaAún no hay calificaciones
- TamiDocumento23 páginasTamikarito cabreraAún no hay calificaciones
- Tamizaje Neonatal CR PDFDocumento3 páginasTamizaje Neonatal CR PDFdatitoxAún no hay calificaciones
- Tamiz Metabolico NeonatalDocumento3 páginasTamiz Metabolico NeonatalLaura ElenaAún no hay calificaciones
- Guias de Práctica Clinica SopDocumento16 páginasGuias de Práctica Clinica SopAntonio SaavedraAún no hay calificaciones
- 6A - GRUPO - 3 - CC - Hipertiroidismo NeonatalDocumento9 páginas6A - GRUPO - 3 - CC - Hipertiroidismo NeonatalLiz GarciaAún no hay calificaciones
- Tamiz Neonatal AmpliadoDocumento19 páginasTamiz Neonatal AmpliadoKarenina NerakAún no hay calificaciones
- ColestasisDocumento50 páginasColestasisomar victorAún no hay calificaciones
- Tamizaje NeonatalDocumento3 páginasTamizaje Neonatalkg972401Aún no hay calificaciones
- Hipotiroidisimo e HiperDocumento9 páginasHipotiroidisimo e HiperAlma Julieta BazánAún no hay calificaciones
- Ginecologia Pediatrica y Adolescente 24082023Documento6 páginasGinecologia Pediatrica y Adolescente 24082023jonathan.perez9849Aún no hay calificaciones
- Tamizaje NeonatalDocumento67 páginasTamizaje NeonatalVeronica CentenoAún no hay calificaciones
- Buen RepasoDocumento197 páginasBuen Repasonetinho210Aún no hay calificaciones
- Hipertiroidismo Neonatal: Esa Redonda Alteraciones Endocrinológicas PerinatalesDocumento5 páginasHipertiroidismo Neonatal: Esa Redonda Alteraciones Endocrinológicas PerinatalesDr Flavio GuillénAún no hay calificaciones
- Sindrome de Ovarios PoliquisticosDocumento48 páginasSindrome de Ovarios PoliquisticosGrecia Torres ValderramaAún no hay calificaciones
- Sindrome Ovario PoliquisticoDocumento14 páginasSindrome Ovario PoliquisticojesusAún no hay calificaciones
- FisiopatologíaDocumento8 páginasFisiopatologíaLiam VergaraAún no hay calificaciones
- Deber 1 - TamizajeDocumento4 páginasDeber 1 - TamizajeIVONNE SCARLET MORANTE ROSEROAún no hay calificaciones
- Conclusiones de Trastornos MetabolicosDocumento8 páginasConclusiones de Trastornos MetabolicosKatherine Altamirano AguilarAún no hay calificaciones
- Valoracionendocrinapropedeuticaii 091117154403 Phpapp01Documento22 páginasValoracionendocrinapropedeuticaii 091117154403 Phpapp01Gerald CrespinAún no hay calificaciones
- FisiopatologíaDocumento6 páginasFisiopatologíaotonooAún no hay calificaciones
- Tiroides y EmbarazoDocumento31 páginasTiroides y EmbarazoSilvia Bermúdez SánchezAún no hay calificaciones
- Hipotiroidismo CongénitoDocumento4 páginasHipotiroidismo CongénitoBrandon UchihaAún no hay calificaciones
- Hipoglicemia NeonatalDocumento15 páginasHipoglicemia Neonatalejleon91Aún no hay calificaciones
- Guías de Diagnóstico y Tratamiento en Neonatología 2006Documento4 páginasGuías de Diagnóstico y Tratamiento en Neonatología 2006Elena DjfAún no hay calificaciones
- Colestasis IntrahepáticaDocumento11 páginasColestasis IntrahepáticaJose Ivan100% (1)
- Seminario de Hiperemesis Gravidica y Enfermedad Trofoblastica GestacionalDocumento50 páginasSeminario de Hiperemesis Gravidica y Enfermedad Trofoblastica GestacionalRoxy ScAún no hay calificaciones
- Hipotiroidismo y EmbarazoDocumento6 páginasHipotiroidismo y EmbarazoCarolina RiveraAún no hay calificaciones
- 4 TiroidesDocumento29 páginas4 TiroidesLido Cardenas VargasAún no hay calificaciones
- SOPDocumento4 páginasSOPMiguel Correa CotuáAún no hay calificaciones
- Síndrome de Ovarios PoliquísticosDocumento25 páginasSíndrome de Ovarios PoliquísticosOmar OrtizAún no hay calificaciones
- ExamenDocumento20 páginasExamenRita ZavaletaAún no hay calificaciones
- Conferencia Diabetes Y EmbarazoDocumento22 páginasConferencia Diabetes Y EmbarazoPaolo ManfrediAún no hay calificaciones
- Guía Clase Hipo-HipertiroidismoDocumento10 páginasGuía Clase Hipo-HipertiroidismoFrancisco CamposAún no hay calificaciones
- Pesquisa NeonatalDocumento15 páginasPesquisa NeonatalRafaella IwamotoAún no hay calificaciones
- Hipoglicemia e Hipocalcemia NeonatalDocumento16 páginasHipoglicemia e Hipocalcemia NeonatalYoselyn Sanchez Sanchez100% (1)
- GPC Hipotiroidismo Congénito en El Primer Nivel de Atención 2015Documento8 páginasGPC Hipotiroidismo Congénito en El Primer Nivel de Atención 2015Cesar Alexis Trejo RoqueAún no hay calificaciones
- Galactorreaok 120929052030 Phpapp01Documento49 páginasGalactorreaok 120929052030 Phpapp01EstefAún no hay calificaciones
- SomatropinaDocumento4 páginasSomatropinaDon ProAún no hay calificaciones
- Sindrome de Ovario PoliquisticoDocumento7 páginasSindrome de Ovario PoliquisticoSamuel Cruz ValdrramaAún no hay calificaciones
- Revision Bibliografica - ClimaterioDocumento9 páginasRevision Bibliografica - ClimaterioTania MonsalvaAún no hay calificaciones
- Anovulacion PDFDocumento15 páginasAnovulacion PDFMagic_OverAún no hay calificaciones
- Sindrome de Ovario PoliquisticoDocumento3 páginasSindrome de Ovario PoliquisticoRosana LujanAún no hay calificaciones
- Endocrinologia CtoDocumento26 páginasEndocrinologia CtoAdriana NavaAún no hay calificaciones
- Tamizaje NeonatalDocumento22 páginasTamizaje NeonatalCristhian Agustin ParedesAún no hay calificaciones
- Errores Congenitos Del Metabolismo y Otros Trastornos GeneticosDocumento54 páginasErrores Congenitos Del Metabolismo y Otros Trastornos Geneticosraul_murillo_5Aún no hay calificaciones
- Hiperglucemia / Hipoglucemia NeonatalDocumento37 páginasHiperglucemia / Hipoglucemia Neonatalmc20temd7939Aún no hay calificaciones
- Hiperemesis GravídicaDocumento20 páginasHiperemesis GravídicaLucianoNeruda100% (1)
- Tirosinemia Tipo 1Documento4 páginasTirosinemia Tipo 1Dario Quimis SantanaAún no hay calificaciones
- Hiperémesis Gravídica Ci1Documento8 páginasHiperémesis Gravídica Ci1Andrea VBAún no hay calificaciones
- Errores Innatos Del MetabolismoDocumento69 páginasErrores Innatos Del MetabolismoCecilia Acosta100% (1)
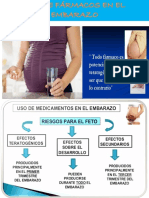
- Uso de Farmacos en El EmbarazoDocumento26 páginasUso de Farmacos en El EmbarazoNiinosk PáizAún no hay calificaciones
- T13 Síndrome de Ovario PoliquísticoDocumento2 páginasT13 Síndrome de Ovario PoliquísticoRodrigo Pérez CuelloAún no hay calificaciones
- Sindrome de Ovario PoliquisticoDocumento7 páginasSindrome de Ovario Poliquisticojuan pablo beltranAún no hay calificaciones
- Taller de HipotiroidismoDocumento16 páginasTaller de HipotiroidismopiloricoAún no hay calificaciones
- Screnning Neonatal y LactanciaDocumento5 páginasScrenning Neonatal y LactanciaGaby EveAún no hay calificaciones
- Enfermedades Hepáticas Propias Del EmbarazoDocumento12 páginasEnfermedades Hepáticas Propias Del EmbarazoMaribel Martinez AchacolloAún no hay calificaciones
- TIROIDOPATIASDocumento17 páginasTIROIDOPATIASDanny FernandezAún no hay calificaciones
- Sangrado Uterino AnormalDocumento10 páginasSangrado Uterino AnormalVictoria FabiAún no hay calificaciones
- EMERGENTODocumento4 páginasEMERGENTOVictoria FabiAún no hay calificaciones
- Anemia y NutricionDocumento7 páginasAnemia y NutricionVictoria FabiAún no hay calificaciones
- Cardiología PediátricaDocumento14 páginasCardiología PediátricaVictoria FabiAún no hay calificaciones
- Control Niño SanoDocumento12 páginasControl Niño SanoVictoria FabiAún no hay calificaciones
- Cuadro Infecciones SexualesDocumento7 páginasCuadro Infecciones SexualesVictoria FabiAún no hay calificaciones
- ACNÉDocumento5 páginasACNÉVictoria FabiAún no hay calificaciones
- Clase GastroDocumento3 páginasClase GastroVictoria FabiAún no hay calificaciones
- GINECOLOGÍADocumento40 páginasGINECOLOGÍAVictoria FabiAún no hay calificaciones
- Control PrenatalDocumento2 páginasControl PrenatalVictoria FabiAún no hay calificaciones
- SÍNCOPEDocumento1 páginaSÍNCOPEVictoria FabiAún no hay calificaciones
- Seguimiento y Correctores de Portfolios: Lucia DiomediDocumento2 páginasSeguimiento y Correctores de Portfolios: Lucia DiomediVictoria FabiAún no hay calificaciones
- Listado de Temas de Clínica Pediátrica y Evaluaciones23Documento4 páginasListado de Temas de Clínica Pediátrica y Evaluaciones23Victoria FabiAún no hay calificaciones
- Enfermedad Celíaca en NiñosDocumento7 páginasEnfermedad Celíaca en NiñosVictoria FabiAún no hay calificaciones
- AcademicaypracticaDocumento2 páginasAcademicaypracticaVictoria FabiAún no hay calificaciones
- Bibliografia Complementaria Aguirre Aspectos Socioantropologicos de La Obesidad en La PobrezaDocumento13 páginasBibliografia Complementaria Aguirre Aspectos Socioantropologicos de La Obesidad en La PobrezaVictoria FabiAún no hay calificaciones
- Diarrea CrónicaDocumento12 páginasDiarrea CrónicaVictoria FabiAún no hay calificaciones
- Cardiología Infantil - ClaseDocumento4 páginasCardiología Infantil - ClaseVictoria FabiAún no hay calificaciones
- Dermatitis AtópicaDocumento5 páginasDermatitis AtópicaVictoria FabiAún no hay calificaciones
- Trabajo Práctico La Atmosfera y El Origen de La VidaDocumento3 páginasTrabajo Práctico La Atmosfera y El Origen de La VidaVictoria FabiAún no hay calificaciones
- La Invaginación Intestinal Se Produce Cuando Una Porción Del Tubo Digestivo Se Introduce Dentro de Otro SegmentoDocumento3 páginasLa Invaginación Intestinal Se Produce Cuando Una Porción Del Tubo Digestivo Se Introduce Dentro de Otro SegmentoVictoria FabiAún no hay calificaciones
- Resumen Romano 3er ParcialDocumento31 páginasResumen Romano 3er ParcialVictoria FabiAún no hay calificaciones
- Libro 1Documento1 páginaLibro 1Victoria FabiAún no hay calificaciones
- Cuadro TP7Documento1 páginaCuadro TP7Victoria FabiAún no hay calificaciones
- Caso Clínico Disparador - Maria Victoria FabiDocumento5 páginasCaso Clínico Disparador - Maria Victoria FabiVictoria FabiAún no hay calificaciones
- Cuadro TP7Documento1 páginaCuadro TP7Victoria FabiAún no hay calificaciones
- Cancer ColorrectalDocumento85 páginasCancer ColorrectalVictoria FabiAún no hay calificaciones
- Portfolio RotacionDocumento1 páginaPortfolio RotacionVictoria FabiAún no hay calificaciones
- Seminario Medio Interno Con AudioDocumento55 páginasSeminario Medio Interno Con AudioVictoria FabiAún no hay calificaciones
- Ca. No Melanoma.Documento2 páginasCa. No Melanoma.Victoria FabiAún no hay calificaciones
- NieblaDocumento2 páginasNieblaMara Dell'AversanaAún no hay calificaciones
- Características de La Comunicación TerapéuticaDocumento2 páginasCaracterísticas de La Comunicación TerapéuticaViviana Malpica100% (2)
- Suficientementebueno PDFDocumento4 páginasSuficientementebueno PDFGabriel Deidara SempaiAún no hay calificaciones
- Informe Cantabria Completo TrujilloDocumento569 páginasInforme Cantabria Completo TrujilloEdu SandovalAún no hay calificaciones
- Investigacion Academica S9 - Tarea - Fichas Textuales y de ResumenDocumento14 páginasInvestigacion Academica S9 - Tarea - Fichas Textuales y de ResumenVictoria Choque FernandezAún no hay calificaciones
- 1 Sanidad Acuicola - IntroduccionDocumento13 páginas1 Sanidad Acuicola - IntroduccionNestor Alejandro Velazco Silva100% (1)
- Circuitos Amplificadores - Amplificador Clase BDocumento6 páginasCircuitos Amplificadores - Amplificador Clase BLuis AndreAún no hay calificaciones
- Matriz de Rotación para Imágenes en VisualDocumento3 páginasMatriz de Rotación para Imágenes en VisualDUVAN FERNANDO PULGARIN MARTINEZAún no hay calificaciones
- Nunca Demasiado Viejo - Angelesoscuros13.PDF Versión 1Documento37 páginasNunca Demasiado Viejo - Angelesoscuros13.PDF Versión 1Thyani JarvisAún no hay calificaciones
- FICHA N°02:: "Diseñamos Una Alternativa de Solución Tecnológica"Documento7 páginasFICHA N°02:: "Diseñamos Una Alternativa de Solución Tecnológica"Juan DiegxAún no hay calificaciones
- RectaDocumento13 páginasRectaCARLOS BERNARDO CAMACHO RIMACHIAún no hay calificaciones
- Presentación Nutrición ParenteralDocumento57 páginasPresentación Nutrición ParenteralJuLie SaLas100% (1)
- Glosarios Primeros AuxiliosDocumento3 páginasGlosarios Primeros AuxiliosSanchez Gamarra AndyAún no hay calificaciones
- Del Diseño PostIndustrial Al MetadiseñoDocumento19 páginasDel Diseño PostIndustrial Al MetadiseñoRicardo Romero0% (1)
- 10 Reglas para Conversar MejorDocumento2 páginas10 Reglas para Conversar MejorVictoria LopezAún no hay calificaciones
- Tarea 3 Geografia FisicaDocumento6 páginasTarea 3 Geografia FisicaLuis veloz50% (2)
- Informe Memoria de Calculo Muros de GravedadDocumento4 páginasInforme Memoria de Calculo Muros de GravedadOscar Luna Pizarro GuerraAún no hay calificaciones
- Resumen Historia de La ProgramaciónDocumento2 páginasResumen Historia de La Programacióneddier de jesus rodriguez mendozaAún no hay calificaciones
- Primer Grado U2 s14Documento4 páginasPrimer Grado U2 s14Rosaria Ponce GonzalesAún no hay calificaciones
- RJ 1992 - 2022oaf-Costo Acdemcios Año 2023Documento4 páginasRJ 1992 - 2022oaf-Costo Acdemcios Año 2023Roberto Fernando CoronadoAún no hay calificaciones
- ST2 Trabajo Tema 3Documento7 páginasST2 Trabajo Tema 3Antonio RuedaAún no hay calificaciones
- Manual Aspel Facture 60 Parte 01Documento19 páginasManual Aspel Facture 60 Parte 01Ricardo CalixtoAún no hay calificaciones
- Sobre Las PasionesDocumento4 páginasSobre Las PasionesIrene GindinAún no hay calificaciones
- Salchichas Chorizos ParrillerosDocumento23 páginasSalchichas Chorizos ParrillerosFreddy Omar Mora Mora100% (1)
- Prueba Sumativa 1Documento1 páginaPrueba Sumativa 1Camila AlonsoAún no hay calificaciones
- 281 Importacion Cuentas Polizas Excel A CFDocumento15 páginas281 Importacion Cuentas Polizas Excel A CFRafael UscangaAún no hay calificaciones
- Soldadura TicDocumento5 páginasSoldadura TicErickAún no hay calificaciones
- Lab 3 Virtual - Ley de FaradayDocumento5 páginasLab 3 Virtual - Ley de Faradaymagaly urregoAún no hay calificaciones
- Ensayo 4Documento3 páginasEnsayo 4Angel RomeroAún no hay calificaciones
- Pueden Los Pensamientos Negativos Cancelar Las Promesas de DiosDocumento2 páginasPueden Los Pensamientos Negativos Cancelar Las Promesas de DiosJhonny Montaño ValdiviaAún no hay calificaciones