También podría gustarte
- Ciencia regulatoria: Medicamentos bio y su relevancia para la saludDe EverandCiencia regulatoria: Medicamentos bio y su relevancia para la saludAún no hay calificaciones
- FDA Regulaciones USDocumento28 páginasFDA Regulaciones USAldo CordobaAún no hay calificaciones
- La necesidad de proteger los datos de prueba de medicamentos biológicos en Ecuador: Análisis comparativoDe EverandLa necesidad de proteger los datos de prueba de medicamentos biológicos en Ecuador: Análisis comparativoAún no hay calificaciones
- Agencias Reguladoras (1) Ing. ClinicaDocumento11 páginasAgencias Reguladoras (1) Ing. Clinicayenifer morenoAún no hay calificaciones
- Regulatory Control - EUDocumento18 páginasRegulatory Control - EUMirra TrejoAún no hay calificaciones
- Insumos Clinicos 5Documento21 páginasInsumos Clinicos 5rayenAún no hay calificaciones
- Resumen 2Documento23 páginasResumen 2DavidAún no hay calificaciones
- Producto de Diagnóstico in VitroDocumento16 páginasProducto de Diagnóstico in Vitrommm777maribelAún no hay calificaciones
- Cap 1 Libro BIO - En.esDocumento17 páginasCap 1 Libro BIO - En.esYONATHAN LENIN HUACASI LUQUEAún no hay calificaciones
- Medical Device Classification - FDADocumento32 páginasMedical Device Classification - FDAMirra TrejoAún no hay calificaciones
- Equipos BomédicosDocumento8 páginasEquipos BomédicosNicoll MarínAún no hay calificaciones
- SELECCIÓN TECNOLOGICADocumento34 páginasSELECCIÓN TECNOLOGICAkelly velandiaAún no hay calificaciones
- Dermatrón (1225) Informe para La CiudadaníaDocumento7 páginasDermatrón (1225) Informe para La CiudadaníaCarlos BenitezAún no hay calificaciones
- Ley de Dispositivos MédicosDocumento15 páginasLey de Dispositivos MédicosMariana0% (1)
- Dispositivos MedicosDocumento19 páginasDispositivos Medicosfisterjesus0% (1)
- Muchas Compañías Fabricantes de Productos Farmacéuticos o de Productos SanitariosDocumento9 páginasMuchas Compañías Fabricantes de Productos Farmacéuticos o de Productos SanitariosAmèrica SalinasAún no hay calificaciones
- Resumenes Cap 3 y 8Documento16 páginasResumenes Cap 3 y 8Natalia Tavera OrozcoAún no hay calificaciones
- Manual Pasantias 2020Documento8 páginasManual Pasantias 2020Astrid JaimesAún no hay calificaciones
- 3.1.8 Suplemento para Dispositivos MedicosDocumento27 páginas3.1.8 Suplemento para Dispositivos MedicosIvan Alvarado OrtegaAún no hay calificaciones
- Normatividad colombiana sobre dispositivos médicosDocumento35 páginasNormatividad colombiana sobre dispositivos médicosYackopo FrapaAún no hay calificaciones
- Guía verificación requisitos equipos biomédicosDocumento9 páginasGuía verificación requisitos equipos biomédicosBetancur AlejandroAún no hay calificaciones
- Normativa Mexicana para biomaterialesDocumento20 páginasNormativa Mexicana para biomaterialesAldo Cordoba100% (1)
- Electro MedicinaDocumento3 páginasElectro MedicinaCastillo AlexanderAún no hay calificaciones
- Bol1 1994Documento4 páginasBol1 1994Bruno PinascoAún no hay calificaciones
- Normas IEC 60601 1Documento9 páginasNormas IEC 60601 1Francisco Javier MontoyaAún no hay calificaciones
- Segunda Tarea de Diseño de Dispositivos MédicosDocumento4 páginasSegunda Tarea de Diseño de Dispositivos Médicosyodalgi oliveroAún no hay calificaciones
- ApuntesDocumento6 páginasApuntesMarina PaucarAún no hay calificaciones
- Manejo eficiente de insumos y dispositivos médicosDocumento19 páginasManejo eficiente de insumos y dispositivos médicosJose RicoAún no hay calificaciones
- INVIMA funciones dispositivos médicos clases riesgoDocumento9 páginasINVIMA funciones dispositivos médicos clases riesgoINGRID TATIANA NIETO ORTIZAún no hay calificaciones
- Pandrh MD Cofepris SpaDocumento23 páginasPandrh MD Cofepris SpaLucia Rodriguez AguilarAún no hay calificaciones
- Tema 12 - Normas - Ley 26.906Documento13 páginasTema 12 - Normas - Ley 26.906Lucia LoperaAún no hay calificaciones
- Resumen Decreto4725Documento13 páginasResumen Decreto4725Diego PeñuelaAún no hay calificaciones
- Guia para Adquisicion de Nueva Tecnologia PDFDocumento10 páginasGuia para Adquisicion de Nueva Tecnologia PDFLuis Fernando Rojas CubillosAún no hay calificaciones
- Tiempo de Vida Promedio de Uso Del Dispositivo MedicoDocumento6 páginasTiempo de Vida Promedio de Uso Del Dispositivo MedicoRosalba ToxquiAún no hay calificaciones
- 01-Respuestas de La Evaluacion Tec-Asuntos RegulatoriosDocumento3 páginas01-Respuestas de La Evaluacion Tec-Asuntos Regulatoriosreclutamientosil89Aún no hay calificaciones
- Clasificacion de Los Equipos Por Riesgo Según InvimaDocumento29 páginasClasificacion de Los Equipos Por Riesgo Según InvimaDianaPaolaPeñuelaAún no hay calificaciones
- OPSHSSMT220024 SpaDocumento15 páginasOPSHSSMT220024 SpaAGUSTIN ENRIQUE SAMPIETROAún no hay calificaciones
- Silvia Perez Regulacion de Dispositivos MedicosDocumento20 páginasSilvia Perez Regulacion de Dispositivos MedicosOPPF DIREMID LORETO100% (1)
- Ord Metrología A Pérez - AndreaDocumento24 páginasOrd Metrología A Pérez - AndreaAlejandrina PerezAún no hay calificaciones
- Ra & Medical DeviceDocumento46 páginasRa & Medical Devicelaura ChicchonAún no hay calificaciones
- La Tecnovigilancia en MexicoDocumento26 páginasLa Tecnovigilancia en MexicoJessy Arroyo100% (1)
- Dispositivos MedicosDocumento3 páginasDispositivos MedicosGabis MuñozAún no hay calificaciones
- Bpm equipos médicos InvimaDocumento11 páginasBpm equipos médicos InvimaAlejo RoseroAún no hay calificaciones
- Clasificacion de Los Dispositivos Medicos Lechuga FernandoDocumento4 páginasClasificacion de Los Dispositivos Medicos Lechuga FernandoFERNANDO RAFAEL LECHUGA MELENDRESAún no hay calificaciones
- Productos MédicosDocumento43 páginasProductos MédicosMarcelo PirizAún no hay calificaciones
- Requisitos para El Registro Sanitario EspañolDocumento3 páginasRequisitos para El Registro Sanitario EspañolheiserosteosintesisAún no hay calificaciones
- Manual de Uso y ReusoDocumento8 páginasManual de Uso y ReusoLuis Miguel TrianaAún no hay calificaciones
- CLASIFICACION y DEFINICIÓN DE DISPOSITIVOS MÉDICASDocumento5 páginasCLASIFICACION y DEFINICIÓN DE DISPOSITIVOS MÉDICASKellin Beltran Ubate50% (2)
- Evaluación Unidad 2Documento7 páginasEvaluación Unidad 2Luis FigueredoAún no hay calificaciones
- Calibracion Equipos BiomedicosDocumento8 páginasCalibracion Equipos BiomedicosDavid JaramilloAún no hay calificaciones
- Norma Técnica 81-2020 Disposiciones Del Drcfpfa Durante El Estado de CalamidadDocumento5 páginasNorma Técnica 81-2020 Disposiciones Del Drcfpfa Durante El Estado de CalamidadRosalino SánchezAún no hay calificaciones
- FDA Regulation of Human Cells and Tissues - SpanishDocumento8 páginasFDA Regulation of Human Cells and Tissues - SpanishViridiana LealAún no hay calificaciones
- Manual de Buenas Prácticas de FarmacovigilanciaDocumento43 páginasManual de Buenas Prácticas de FarmacovigilanciaMiracoli BaViAún no hay calificaciones
- Inst Bioméd clasif dispositivosDocumento27 páginasInst Bioméd clasif dispositivosOthoniel Hernandez OvandoAún no hay calificaciones
- Clasificacion de Riesgos de Dispositivos MedicosDocumento31 páginasClasificacion de Riesgos de Dispositivos MedicosAlejandro Ramirez75% (8)
- Manual de tecnovigilancia_1 (1) (8)Documento13 páginasManual de tecnovigilancia_1 (1) (8)emi.biomedicos.ejecafeteroAún no hay calificaciones
- Marco Legal y Requerimientos para La Homologación deDocumento18 páginasMarco Legal y Requerimientos para La Homologación deERLYN JULIÁN CEDIEL SÁNCHEZAún no hay calificaciones
- Clasificación de riesgo DMDocumento21 páginasClasificación de riesgo DMAlejandra Ramirez SanchezAún no hay calificaciones
- Clasificacion Dispositivos MedicosDocumento4 páginasClasificacion Dispositivos MedicosYolyAún no hay calificaciones
- Reactivo Vigilancia Decreto 3770 de 2004 y Resolucion 132 de 2006Documento14 páginasReactivo Vigilancia Decreto 3770 de 2004 y Resolucion 132 de 2006Greisy Tapia SAún no hay calificaciones
- EXAMEN PARCIAL 1A - Revisión Del IntentoDocumento8 páginasEXAMEN PARCIAL 1A - Revisión Del IntentofranAún no hay calificaciones
- Circuitos OperacinalesDocumento9 páginasCircuitos OperacinalesfranAún no hay calificaciones
- Lab02 - CPUDocumento21 páginasLab02 - CPUfranAún no hay calificaciones
- PLC y DCSDocumento17 páginasPLC y DCSfranAún no hay calificaciones
- Sistemas de Telecomunicaciones - Práctica 6Documento10 páginasSistemas de Telecomunicaciones - Práctica 6franAún no hay calificaciones
- Analisis de La Realidad PeruanaDocumento3 páginasAnalisis de La Realidad PeruanafranAún no hay calificaciones
- SOLUCION PRUEBA DE PRUEBA DE ENTRADA GRUPO C SIyB 2022 - ADocumento1 páginaSOLUCION PRUEBA DE PRUEBA DE ENTRADA GRUPO C SIyB 2022 - AfranAún no hay calificaciones
- Lab01 - Serie y Modelos de PLCDocumento18 páginasLab01 - Serie y Modelos de PLCfranAún no hay calificaciones
- Expo Biomedicas Tema 2Documento71 páginasExpo Biomedicas Tema 2franAún no hay calificaciones
- Motores y generadores: funcionamiento y principios básicosDocumento26 páginasMotores y generadores: funcionamiento y principios básicosfranAún no hay calificaciones
- Protocolos de Comunicación OSIDocumento20 páginasProtocolos de Comunicación OSIfranAún no hay calificaciones
- Ensayo de Comunicacion VirtualDocumento3 páginasEnsayo de Comunicacion VirtualfranAún no hay calificaciones
- Laboratorio Instalaciones ElectricasDocumento2 páginasLaboratorio Instalaciones ElectricasfranAún no hay calificaciones
- Grafcet MergedDocumento315 páginasGrafcet Mergedfran100% (1)
- Ejercicios de funciones, vectores y problemas de población y costosDocumento1 páginaEjercicios de funciones, vectores y problemas de población y costosfranAún no hay calificaciones
- Circuitos ElectrónicosDocumento8 páginasCircuitos ElectrónicosfranAún no hay calificaciones
- SolDocumento6 páginasSolBryam SiavichayAún no hay calificaciones
- Ejercicios Razonamiento MatemáticoDocumento1 páginaEjercicios Razonamiento MatemáticofranAún no hay calificaciones
- Ejercicios de PotenciaDocumento1 páginaEjercicios de PotenciafranAún no hay calificaciones
- Ejercicios Razonamiento MatemáticoDocumento1 páginaEjercicios Razonamiento MatemáticofranAún no hay calificaciones
- Ejercicios LógicosDocumento3 páginasEjercicios LógicosfranAún no hay calificaciones
- Ejercicios de PotenciaDocumento1 páginaEjercicios de PotenciafranAún no hay calificaciones
- Cultura y RazaDocumento5 páginasCultura y RazafranAún no hay calificaciones
- Practica MateDocumento9 páginasPractica MatefranAún no hay calificaciones
- Ejercicios Máquinas ElectricasDocumento2 páginasEjercicios Máquinas ElectricasfranAún no hay calificaciones
- Ecuaciones y SusesionesDocumento4 páginasEcuaciones y SusesionesfranAún no hay calificaciones
- Ejercicios DiodosDocumento2 páginasEjercicios DiodosfranAún no hay calificaciones
- Ejercicios de álgebra, ecuaciones y problemasDocumento1 páginaEjercicios de álgebra, ecuaciones y problemasfranAún no hay calificaciones
- Metodo CornellDocumento6 páginasMetodo CornellfranAún no hay calificaciones
- Electronica de PotenciaDocumento12 páginasElectronica de PotenciaEdwin MurciaAún no hay calificaciones
- Solucionario Examen de Admision San Marcos 2017 1 37466 Downloable 1636111Documento112 páginasSolucionario Examen de Admision San Marcos 2017 1 37466 Downloable 1636111PAUL ANDRES CHACHA100% (1)
- Mapa Conceptual. de La Auditoria InternaDocumento1 páginaMapa Conceptual. de La Auditoria InternaMaira Alejandra Blanco Orjuela0% (1)
- Proyecto FencytDocumento7 páginasProyecto FencytRoyger Orbe FloresAún no hay calificaciones
- Guía 3 InfografíaDocumento2 páginasGuía 3 Infografíafarid ibañez gutierrezAún no hay calificaciones
- Antep Roy EctoDocumento12 páginasAntep Roy EctoPOLO-FIIIDT CIMECDIAún no hay calificaciones
- Universidad Autónoma de Nicaragua ordena contactosDocumento11 páginasUniversidad Autónoma de Nicaragua ordena contactosLuisAún no hay calificaciones
- PDF DR-C240Documento4 páginasPDF DR-C240Orlando Valderrama HernandezAún no hay calificaciones
- Si SE PUEDE USAR AUDIFONOS MEDICOS PARA TRABAJAR EN MINA Y PLANTA SEGUN OHSAS 18001Documento5 páginasSi SE PUEDE USAR AUDIFONOS MEDICOS PARA TRABAJAR EN MINA Y PLANTA SEGUN OHSAS 18001cuarzo100% (1)
- Laboratorio. 4 Codigo G02 - G03 - Simulación - AsdDocumento20 páginasLaboratorio. 4 Codigo G02 - G03 - Simulación - AsdAngelAún no hay calificaciones
- InformaticaDocumento55 páginasInformaticaGabriela Peralta Gabi BolsonAún no hay calificaciones
- SogoDocumento3 páginasSogoAugusto gonzalezAún no hay calificaciones
- Diagramas de ClasesDocumento8 páginasDiagramas de ClasesDragonrlsDiosAún no hay calificaciones
- Delito Abuso Infantil, GroomingDocumento59 páginasDelito Abuso Infantil, Groomingmayth.victorioAún no hay calificaciones
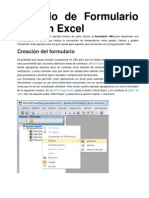
- Ejemplo de Formulario VBA en ExcelDocumento6 páginasEjemplo de Formulario VBA en ExcelEdson LópezAún no hay calificaciones
- D 2020 003 Directivas para SupervisionDocumento44 páginasD 2020 003 Directivas para Supervisionrjcc copareAún no hay calificaciones
- Taller 1 ControlDocumento29 páginasTaller 1 ControlCamilo Alberto Marin PizzaAún no hay calificaciones
- 11 Ejercicios para Dominar El TecladoDocumento12 páginas11 Ejercicios para Dominar El TecladoCarlos Fernando Aguayo CárdenasAún no hay calificaciones
- Páginas DesdeAPI RP 7G-2 TraducidoDocumento1 páginaPáginas DesdeAPI RP 7G-2 TraducidoGarcia C L Alberto100% (1)
- 1.0.1.2 PC Septup For WorkshopDocumento7 páginas1.0.1.2 PC Septup For WorkshopJessica Carolina CalapaquiAún no hay calificaciones
- Proyecto La GranjaDocumento27 páginasProyecto La GranjaRosana AstaizaAún no hay calificaciones
- Diseño - Imagen CorporativaDocumento18 páginasDiseño - Imagen CorporativaLAURA MELISSA CARDENAS TALEROAún no hay calificaciones
- Redes-LAN-WANDocumento1 páginaRedes-LAN-WANPablo Israel Bolaños PárragaAún no hay calificaciones
- Cantos Sagrados Mbya Guarani PDFDocumento13 páginasCantos Sagrados Mbya Guarani PDFTam MuroAún no hay calificaciones
- UPS TecnologiaDocumento1 páginaUPS TecnologiaCarlosViñanzacaAún no hay calificaciones
- MONT IonizadorDocumento3 páginasMONT IonizadorConde JuanjoAún no hay calificaciones
- Ensayo Sistemas SigDocumento4 páginasEnsayo Sistemas SigYojan Chaverra MenaAún no hay calificaciones
- La Ley de Moore y La Ley de MetcalfeDocumento2 páginasLa Ley de Moore y La Ley de MetcalfeAlejandroAún no hay calificaciones
- Examen DiagnosticoDocumento3 páginasExamen DiagnosticoFiliberto PadillaAún no hay calificaciones
- Informe IBM Banca Multicanal 2012Documento72 páginasInforme IBM Banca Multicanal 2012Francisco Antonio Álvarez CanoAún no hay calificaciones
- Fisiopatología de las enfermedades cardiovascularesDe EverandFisiopatología de las enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)
- Terapia cognitiva: Conceptos básicos y profundizaciónDe EverandTerapia cognitiva: Conceptos básicos y profundizaciónCalificación: 5 de 5 estrellas5/5 (1)
- TDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosDe EverandTDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosCalificación: 4 de 5 estrellas4/5 (8)
- Psiconeuroinmunología para la práctica clínicaDe EverandPsiconeuroinmunología para la práctica clínicaCalificación: 5 de 5 estrellas5/5 (4)
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- Ansiedad infantil. Los trastornos explicados a los padresDe EverandAnsiedad infantil. Los trastornos explicados a los padresCalificación: 4.5 de 5 estrellas4.5/5 (25)
- El libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)De EverandEl libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)Calificación: 3 de 5 estrellas3/5 (2)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Genética general: Libro de textoDe EverandGenética general: Libro de textoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Trauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesDe EverandTrauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesCalificación: 1 de 5 estrellas1/5 (1)
- Manual para la administración de medicamentos desde el proceso de atención de enfermería: Un enfoque para la seguridad del pacienteDe EverandManual para la administración de medicamentos desde el proceso de atención de enfermería: Un enfoque para la seguridad del pacienteCalificación: 2.5 de 5 estrellas2.5/5 (4)
- Puntos gatillo y puntos acupunturales (Color)De EverandPuntos gatillo y puntos acupunturales (Color)Calificación: 4.5 de 5 estrellas4.5/5 (13)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- El autismo: Reflexiones y pautas para comprenderlo y abordarloDe EverandEl autismo: Reflexiones y pautas para comprenderlo y abordarloCalificación: 4 de 5 estrellas4/5 (7)
- El concepto Mulligan de terapia manual (Color)De EverandEl concepto Mulligan de terapia manual (Color)Calificación: 5 de 5 estrellas5/5 (3)
- Póngase En Forma Sin Salir De Su CasaDe EverandPóngase En Forma Sin Salir De Su CasaCalificación: 4.5 de 5 estrellas4.5/5 (4)
- Prescripción de ejercico físico para la saludDe EverandPrescripción de ejercico físico para la saludCalificación: 5 de 5 estrellas5/5 (1)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Fundamentos de medicina tradicional chinaDe EverandFundamentos de medicina tradicional chinaCalificación: 4.5 de 5 estrellas4.5/5 (5)
- Terapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaDe EverandTerapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Puntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)De EverandPuntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)Calificación: 4.5 de 5 estrellas4.5/5 (23)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)