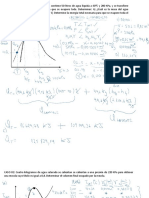
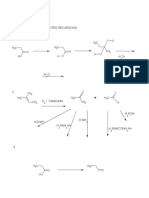
También podría gustarte
- UF1357 - Regeneración óptima de los alimentosDe EverandUF1357 - Regeneración óptima de los alimentosCalificación: 5 de 5 estrellas5/5 (1)
- Acondicionado de camas, prendas de vestir y ropa de hogar. SSCI0109De EverandAcondicionado de camas, prendas de vestir y ropa de hogar. SSCI0109Aún no hay calificaciones
- Analisis de Harinas - TatiDocumento5 páginasAnalisis de Harinas - TatiKaren SoledadAún no hay calificaciones
- Introduccion Al Aire AcondicionadoDocumento66 páginasIntroduccion Al Aire Acondicionadoelmer chañi100% (2)
- Informe de Determinación de Humedad y CenizasDocumento14 páginasInforme de Determinación de Humedad y CenizasLucaps Acedo Kiroma88% (8)
- Informe 2 - Humedad y CenizasDocumento6 páginasInforme 2 - Humedad y CenizasAngie CampoverdeAún no hay calificaciones
- Akemy BalanceDocumento11 páginasAkemy BalanceAlanis MayuriAún no hay calificaciones
- Tecnología de las conservas de frutas y vegetales. Segunda parteDe EverandTecnología de las conservas de frutas y vegetales. Segunda parteAún no hay calificaciones
- Análisis de Cereales y HarinasDocumento18 páginasAnálisis de Cereales y HarinasAna Liceth CasillaAún no hay calificaciones
- Determinacion de Cenizas y Humedad-1Documento3 páginasDeterminacion de Cenizas y Humedad-1Miguel angel Burgoa carbajal100% (2)
- Informe 1. Análisis de La Composición y Calidad Fisicoquímica de Los AlimentosDocumento9 páginasInforme 1. Análisis de La Composición y Calidad Fisicoquímica de Los AlimentosJorge ForeroAún no hay calificaciones
- PRACTICA N Analisis ProximalDocumento12 páginasPRACTICA N Analisis Proximalleydi ninaAún no hay calificaciones
- Layout IdealizadoDocumento15 páginasLayout IdealizadoEvelin Cuevas MoralesAún no hay calificaciones
- Cereales industria 40Documento23 páginasCereales industria 40Lhu IZAún no hay calificaciones
- Aplicación Industrial: Determinación de Materia Grasa y Acidez de La Materia GrasaDocumento9 páginasAplicación Industrial: Determinación de Materia Grasa y Acidez de La Materia GrasaXimee SoriaAún no hay calificaciones
- LABORATORIO # 6 CerealesDocumento26 páginasLABORATORIO # 6 CerealesAbigail Vidal BaldelomarAún no hay calificaciones
- Practica N°4Documento8 páginasPractica N°4Yunni Alixon0% (1)
- Análisis Oficial OfiDocumento25 páginasAnálisis Oficial OfiRichard MurilloAún no hay calificaciones
- Análisis químico proximal de alimentosDocumento11 páginasAnálisis químico proximal de alimentosJuan Agustin Cuadra SotoAún no hay calificaciones
- Análisis Oficial1.1 PROYECTO 1Documento25 páginasAnálisis Oficial1.1 PROYECTO 1Nain josue CardozoAún no hay calificaciones
- PRACTICA #1proceso de Elaboración de La Mermelada VersiónDocumento12 páginasPRACTICA #1proceso de Elaboración de La Mermelada Versióntransportesrosa2020Aún no hay calificaciones
- Analisis Proximal UAMDocumento19 páginasAnalisis Proximal UAMricharrwayneAún no hay calificaciones
- HarinasDocumento8 páginasHarinasKaren SoledadAún no hay calificaciones
- Informe de CerealesDocumento7 páginasInforme de CerealesJuan Guillermo Mencia SuarezAún no hay calificaciones
- Análisis alim Humedad CenizasDocumento4 páginasAnálisis alim Humedad CenizasFabian Sarango RivasAún no hay calificaciones
- TallerDocumento8 páginasTallerKimberly TusaAún no hay calificaciones
- Determinaciòn de Cenizas en Cereal Del Oryza SativaDocumento17 páginasDeterminaciòn de Cenizas en Cereal Del Oryza SativaJuan Carlos Herrera FacundoAún no hay calificaciones
- Determinacion de Cenizas, Humedad y Grasas en La FrutaDocumento5 páginasDeterminacion de Cenizas, Humedad y Grasas en La FrutaBlankita QuiraAún no hay calificaciones
- Determinacion de LipidosDocumento16 páginasDeterminacion de LipidosAna Isabel Ruiz SanchezAún no hay calificaciones
- NMX RosaDocumento8 páginasNMX RosaRouSs DOominguezAún no hay calificaciones
- MR 123Documento6 páginasMR 123Natalia GuevaraAún no hay calificaciones
- Métodos Instrumentales de Análisis - Harina de MaízDocumento17 páginasMétodos Instrumentales de Análisis - Harina de Maízjoselin vargasAún no hay calificaciones
- PRÁCTICA No 12 2023b Cereales y Determinación de GlutenDocumento5 páginasPRÁCTICA No 12 2023b Cereales y Determinación de GlutenAlana Rodriguez MoralesAún no hay calificaciones
- Dterminacion Mesofilos y HumedadDocumento8 páginasDterminacion Mesofilos y HumedadGABRIELA FAUSTO FRAGOZAAún no hay calificaciones
- Laboratorio de cereales: Control de calidadDocumento10 páginasLaboratorio de cereales: Control de calidadBrandon ParicaguaAún no hay calificaciones
- Determinación de Extracto Etéreo en Los AlimentosDocumento8 páginasDeterminación de Extracto Etéreo en Los AlimentosRosmery Magali Cantani QuispeAún no hay calificaciones
- Determinación extracto étereo alimentosDocumento8 páginasDeterminación extracto étereo alimentosRosmery Magali Cantani QuispeAún no hay calificaciones
- Determinación de Humedad y CenizasDocumento7 páginasDeterminación de Humedad y Cenizaslisbeth yajaira guerrero mestanzaAún no hay calificaciones
- PRÁCTICA No 12 2023b Cereales y Determinación de GlutenDocumento7 páginasPRÁCTICA No 12 2023b Cereales y Determinación de GlutenAlana Rodriguez MoralesAún no hay calificaciones
- Practicos CerealesDocumento5 páginasPracticos CerealesMaria Eugenia TonelliAún no hay calificaciones
- Práctica #1 Determinación de HumedadDocumento8 páginasPráctica #1 Determinación de HumedadNahilyn VillanuevaAún no hay calificaciones
- Clase 4 Comparacion y CenizasDocumento23 páginasClase 4 Comparacion y CenizasSilvana Torres GallegosAún no hay calificaciones
- Analisis de CerealesDocumento9 páginasAnalisis de CerealeskejolaolAún no hay calificaciones
- ANÁLISIS DE HARINASDocumento9 páginasANÁLISIS DE HARINASFiorella SotoAún no hay calificaciones
- Determinacion de AlimentosDocumento9 páginasDeterminacion de AlimentosLlocclla Ccasani GloriaAún no hay calificaciones
- Lab de Teleche 2Documento55 páginasLab de Teleche 2Natali BalboaAún no hay calificaciones
- Metodo de DetergenteDocumento5 páginasMetodo de DetergenteROSMERYAún no hay calificaciones
- Analisis Bromatologico (Valeria)Documento26 páginasAnalisis Bromatologico (Valeria)sheldomAún no hay calificaciones
- Lab #3 - Grasa Cruda TotalDocumento5 páginasLab #3 - Grasa Cruda TotalMaria A MoralesAún no hay calificaciones
- Determinacion de HumedadDocumento13 páginasDeterminacion de HumedadCarlos CanchisAún no hay calificaciones
- Practica 1. Determinacion de HumedadDocumento5 páginasPractica 1. Determinacion de HumedadCamila solanoAún no hay calificaciones
- Lab. de Tec. Alimentos I.doc-1Documento91 páginasLab. de Tec. Alimentos I.doc-1Lizet TincutaAún no hay calificaciones
- NCh0101 1981 PDFDocumento13 páginasNCh0101 1981 PDFErick Anddre Briones CaceresAún no hay calificaciones
- GPL 1 Bioquímica Agroindustrial Hidrólisis de AlmidonDocumento5 páginasGPL 1 Bioquímica Agroindustrial Hidrólisis de AlmidonYolanda SalazarAún no hay calificaciones
- Practica 1 - Extraccion Por SolventesDocumento15 páginasPractica 1 - Extraccion Por SolventesGabriela Ticona Luque100% (1)
- Lab - Tec de La LecheDocumento71 páginasLab - Tec de La LecheRocio Andrea CasillaAún no hay calificaciones
- Determinacion de GrasaDocumento8 páginasDeterminacion de GrasaThony Arqueño YllatopaAún no hay calificaciones
- BromatologiaDocumento19 páginasBromatologiaAngel OsorioAún no hay calificaciones
- Laboratorio #7Documento19 páginasLaboratorio #7Kelin RuelasAún no hay calificaciones
- Practica N°1 de Aceites y Grasas PDFDocumento25 páginasPractica N°1 de Aceites y Grasas PDFKarenth Murillo ChoqueAún no hay calificaciones
- Análisis Físico Químico de Los CerealesDocumento26 páginasAnálisis Físico Químico de Los CerealesMalü MaldonadoAún no hay calificaciones
- Laboratorios Alimentación Animal A P MDocumento4 páginasLaboratorios Alimentación Animal A P MANNABEL INES POBLETE MAYORGAAún no hay calificaciones
- Quimica de Los AlimentosDocumento9 páginasQuimica de Los AlimentosEvelin Cuevas MoralesAún no hay calificaciones
- Proteina MicrobianaDocumento13 páginasProteina MicrobianaEvelin Cuevas MoralesAún no hay calificaciones
- Fermentacion de Aminoacidos - Biotecnologia ExpoDocumento15 páginasFermentacion de Aminoacidos - Biotecnologia ExpoEvelin Cuevas MoralesAún no hay calificaciones
- Ciclo del azufre y sus funciones vitales enDocumento14 páginasCiclo del azufre y sus funciones vitales enEvelin Cuevas MoralesAún no hay calificaciones
- Calor y vaporización de agua: 3 casos de transferencia térmica y cambios de estadoDocumento4 páginasCalor y vaporización de agua: 3 casos de transferencia térmica y cambios de estadoEvelin Cuevas MoralesAún no hay calificaciones
- Industria de Los EspumasDocumento21 páginasIndustria de Los EspumasEvelin Cuevas MoralesAún no hay calificaciones
- Exposicion 1 - Gestion de ProduccionDocumento12 páginasExposicion 1 - Gestion de ProduccionEvelin Cuevas MoralesAún no hay calificaciones
- Fermentación AlcoholicaDocumento20 páginasFermentación AlcoholicaEvelin Cuevas MoralesAún no hay calificaciones
- Presentacion Encuentra Tu Creatividad Papel AzulDocumento16 páginasPresentacion Encuentra Tu Creatividad Papel AzulEvelin Cuevas MoralesAún no hay calificaciones
- Universidad Nacional: Marco Teórico de la InvestigaciónDocumento60 páginasUniversidad Nacional: Marco Teórico de la InvestigaciónEvelin Cuevas MoralesAún no hay calificaciones
- Herramientas BasicasDocumento33 páginasHerramientas BasicasEvelin Cuevas MoralesAún no hay calificaciones
- Paiche en Conserva Ok OkDocumento104 páginasPaiche en Conserva Ok OkEvelin Cuevas MoralesAún no hay calificaciones
- Medición y organización del trabajo en la industria alimentariaDocumento36 páginasMedición y organización del trabajo en la industria alimentariaEvelin Cuevas MoralesAún no hay calificaciones
- BPM - Panaderia DoroteoDocumento58 páginasBPM - Panaderia DoroteoEvelin Cuevas MoralesAún no hay calificaciones
- POLARIMETRÍADocumento4 páginasPOLARIMETRÍAAriana Ulate67% (3)
- Proceso flotación Cu-Zn UNTDocumento12 páginasProceso flotación Cu-Zn UNTWilliam Anthony Chapoñan CajusolAún no hay calificaciones
- Bioisosterismo PDFDocumento18 páginasBioisosterismo PDFAlvaro Turizo0% (1)
- Tinciones Hematologicas (Panoptico Rapido)Documento6 páginasTinciones Hematologicas (Panoptico Rapido)Luna Gabriela100% (1)
- Halógenos 2021Documento29 páginasHalógenos 2021Nicolas Emanuel CalvoAún no hay calificaciones
- Ejercicios Geometria (Brown)Documento2 páginasEjercicios Geometria (Brown)tratame bonito que es mi primera vezAún no hay calificaciones
- Nomenclatura y Formulación de Compuestos Inorgánicos - Di PrinzioDocumento15 páginasNomenclatura y Formulación de Compuestos Inorgánicos - Di Prinziosantivalenarias22Aún no hay calificaciones
- Clasificación de Laboratorios, Material y ReactivosDocumento17 páginasClasificación de Laboratorios, Material y ReactivosMiriam SalasAún no hay calificaciones
- Informe de Quimica AnaliticaDocumento24 páginasInforme de Quimica AnaliticaDarlene QuispeAún no hay calificaciones
- Malla 8Documento12 páginasMalla 8Adrián jose Jiménez guevaraAún no hay calificaciones
- (E) Resumen Capítulo 77Documento8 páginas(E) Resumen Capítulo 77aleAún no hay calificaciones
- Calculos Diluciones DesinfectantesDocumento2 páginasCalculos Diluciones DesinfectantesAndrés YépezAún no hay calificaciones
- Jorge Monsalve Tarea4Documento7 páginasJorge Monsalve Tarea4Jorge Monsalve SalazarAún no hay calificaciones
- Guía de Estudio sobre Teorías Ácido-BaseDocumento8 páginasGuía de Estudio sobre Teorías Ácido-BaseSonya BarriosAún no hay calificaciones
- Celdas fotoelectroquímicas para producción de hidrógenoDocumento33 páginasCeldas fotoelectroquímicas para producción de hidrógenoMichael Abre BouzAún no hay calificaciones
- Reaccion de Diels-Alder - Equipo-3 - 4IM54Documento23 páginasReaccion de Diels-Alder - Equipo-3 - 4IM54kkrotoAún no hay calificaciones
- Practica y Taller Quimicos PDFDocumento3 páginasPractica y Taller Quimicos PDFbryan0% (2)
- Transferencia de Masa CompletarDocumento17 páginasTransferencia de Masa CompletarWilliam Vargas CastroAún no hay calificaciones
- 39 - Radiador - Desmontar - y MontarDocumento5 páginas39 - Radiador - Desmontar - y MontarInerAún no hay calificaciones
- Análisis de ProteínasDocumento7 páginasAnálisis de ProteínasJonathan Andres Ortiz ForeroAún no hay calificaciones
- NitritosDocumento10 páginasNitritosValeria Vega RiveraAún no hay calificaciones
- 3° Parcial de Qca Gral Con RtasDocumento1 página3° Parcial de Qca Gral Con RtasEvelin CarpioAún no hay calificaciones
- Deshidratación osmótica de palta fuerteDocumento9 páginasDeshidratación osmótica de palta fuerteKELLY MABEL BRICEÑO PRETELLAún no hay calificaciones
- Guia 1 QuimicaDocumento82 páginasGuia 1 QuimicaSteven DanielAún no hay calificaciones
- Chalcona - Wikipedia, La Enciclopedia LibreDocumento3 páginasChalcona - Wikipedia, La Enciclopedia LibreMateo Arroyave MoralesAún no hay calificaciones
- Practicas de Lab. Q.Orgánica II I.QDocumento34 páginasPracticas de Lab. Q.Orgánica II I.QAndrés Cortés TorresAún no hay calificaciones
- Ficha Tecnica J. MagicoDocumento2 páginasFicha Tecnica J. MagicoJessika VargasAún no hay calificaciones
- TAREA 5. OrganikDocumento6 páginasTAREA 5. OrganikjusellyAún no hay calificaciones