También podría gustarte
- Logica Proposicional TerminadoDocumento5 páginasLogica Proposicional TerminadoCarlos Alberto Rojas PecerosAún no hay calificaciones
- Bujias - Incandescentes Bosch y Reles PrecalentadoresDocumento28 páginasBujias - Incandescentes Bosch y Reles Precalentadorescarlosyakimchuk100% (1)
- Cuestionario Sobre Modelos AtómicosDocumento1 páginaCuestionario Sobre Modelos AtómicosLucre Corral92% (12)
- Normas Tecnicas para Pozos A TierraDocumento9 páginasNormas Tecnicas para Pozos A Tierraangel_goyés75% (4)
- Anemia A Células FalciformesDocumento16 páginasAnemia A Células FalciformesRosana Lluén SiesquénAún no hay calificaciones
- IQ 04 Identificación y Desnaturalización de ProteínasDocumento7 páginasIQ 04 Identificación y Desnaturalización de ProteínasKYRA NAZARETH MOREIRA BERGESAún no hay calificaciones
- Proteinas G4Documento8 páginasProteinas G4ANGYE ISABEL MONGE AVILESAún no hay calificaciones
- Taller BiocompuestosDocumento9 páginasTaller BiocompuestosCamilo AndresAún no hay calificaciones
- Aminoácidos y ProteínasDocumento21 páginasAminoácidos y ProteínasEstefania SaladoAún no hay calificaciones
- Guia Practicos Computacion Proteinas 1 y 2Documento14 páginasGuia Practicos Computacion Proteinas 1 y 2E-f-f-yAún no hay calificaciones
- Quimica 02 PDFDocumento13 páginasQuimica 02 PDFJülîånJPAún no hay calificaciones
- Solucionario Ensayo CB-144 2015 PDFDocumento30 páginasSolucionario Ensayo CB-144 2015 PDFLilia Marianela Salazar SánchezAún no hay calificaciones
- 2017 TP03 Macromoléculas ModificadoDocumento9 páginas2017 TP03 Macromoléculas ModificadonataliaAún no hay calificaciones
- Estruct. 3D Proteinas (APnro7)Documento5 páginasEstruct. 3D Proteinas (APnro7)JL FavAún no hay calificaciones
- Taller BioquimicaDocumento7 páginasTaller BioquimicaNicolas Rodriguez CaviedesAún no hay calificaciones
- Ejercicios GeneticaDocumento6 páginasEjercicios GeneticaEliana Caro100% (1)
- UntitledDocumento12 páginasUntitledJaidibe AckermanAún no hay calificaciones
- Guía de Trabajos Prácticos Y de Ejercicios Curso de Bioquímica para PEMQBDocumento29 páginasGuía de Trabajos Prácticos Y de Ejercicios Curso de Bioquímica para PEMQBMarcelo Campos PalmaAún no hay calificaciones
- Informe Practico N°1 BiologiaDocumento11 páginasInforme Practico N°1 BiologiaMbugAún no hay calificaciones
- Informe 1 Lab BioquimicaDocumento10 páginasInforme 1 Lab BioquimicaCristóbal Ignacio WittersheimAún no hay calificaciones
- Apitulo 4 Estructura y Funcion CelularDocumento5 páginasApitulo 4 Estructura y Funcion CelularBananero RamosAún no hay calificaciones
- Proteínas y Transporte de MembranaDocumento19 páginasProteínas y Transporte de MembranaMARIANA ALVAREZ RESTREPOAún no hay calificaciones
- Isomería de Compuestos InorgánicosDocumento11 páginasIsomería de Compuestos InorgánicosCatalinaAún no hay calificaciones
- Biologia Selectividad Examen 9 Resuelto Castilla y Leon WWW - Siglo21x.blogspotDocumento8 páginasBiologia Selectividad Examen 9 Resuelto Castilla y Leon WWW - Siglo21x.blogspotmonografiasxAún no hay calificaciones
- Bio Sep 03Documento7 páginasBio Sep 03David Del Pozo MerinoAún no hay calificaciones
- Material de Apoyo TP 2 - 3a Parte.-ProteínasDocumento6 páginasMaterial de Apoyo TP 2 - 3a Parte.-Proteínasotavionov9Aún no hay calificaciones
- PROTEINASDocumento10 páginasPROTEINASAngel Luis Tocuyo RivasAún no hay calificaciones
- Taller para ResolverDocumento11 páginasTaller para ResolverReinaldo De la RosaAún no hay calificaciones
- Biologia Evau Jun18Documento9 páginasBiologia Evau Jun18santiagomarlet59Aún no hay calificaciones
- Practica1 PDFDocumento13 páginasPractica1 PDFJesús SaucedoAún no hay calificaciones
- BIOQUIMICA - ResumenDocumento7 páginasBIOQUIMICA - ResumenFri FairAún no hay calificaciones
- UNIVERSIDAD ESTATAL AMAZÓNICA ProteinasDocumento6 páginasUNIVERSIDAD ESTATAL AMAZÓNICA ProteinasSâmmý JmNzAún no hay calificaciones
- Estructuras y Funciòn de Las MacromolèculasDocumento8 páginasEstructuras y Funciòn de Las MacromolèculasCamila YepesAún no hay calificaciones
- Taller Citoesqueletos y Movilidad CelularDocumento4 páginasTaller Citoesqueletos y Movilidad CelularJose CeledonAún no hay calificaciones
- GQGL R33Documento5 páginasGQGL R33LeoAún no hay calificaciones
- Taller 1 Proteinas - Biologia MolecularDocumento8 páginasTaller 1 Proteinas - Biologia MolecularPaola Brito SierraAún no hay calificaciones
- Aislación y Purificación de ProteínasDocumento10 páginasAislación y Purificación de ProteínasDiana Alexandra Santos Rivera100% (1)
- Bioquimica Trabajo MarcelaDocumento5 páginasBioquimica Trabajo Marceladlareg ziurAún no hay calificaciones
- ProteínasDocumento7 páginasProteínasICKER LEONARDO DIAZ AGUILARAún no hay calificaciones
- Facultad de Medicina3Documento5 páginasFacultad de Medicina3johnnie “johnnie el crack” contrerasAún no hay calificaciones
- Cuestionario BioquimicaDocumento22 páginasCuestionario BioquimicaMIREYA HOLGUIN MENDEZAún no hay calificaciones
- Transcripcion Biogenesis de MP y OrganelosDocumento24 páginasTranscripcion Biogenesis de MP y OrganelosJonathan GonzalezAún no hay calificaciones
- Tutoria Investigacion y Defenza Bioquímica Ii 2024Documento13 páginasTutoria Investigacion y Defenza Bioquímica Ii 2024Chema VenegaAún no hay calificaciones
- ProteínasDocumento92 páginasProteínasDafne RiveraAún no hay calificaciones
- Ensayo Semana 2 BioquimicaDocumento5 páginasEnsayo Semana 2 BioquimicaJohn RosalesAún no hay calificaciones
- Introducción A La Biología Guia TPDocumento11 páginasIntroducción A La Biología Guia TPGonzalo PereyraAún no hay calificaciones
- Preinforme 6 - Química OrgánicaDocumento23 páginasPreinforme 6 - Química OrgánicaCriadero San FransiscoAún no hay calificaciones
- Isomeria UruguayeducaDocumento9 páginasIsomeria UruguayeducaAngélica R.Aún no hay calificaciones
- Preguntas II Parcial BioquímicaDocumento5 páginasPreguntas II Parcial BioquímicaSaid AlejandroAún no hay calificaciones
- Entrega 1. HemoglobinaDocumento6 páginasEntrega 1. HemoglobinaISABELLA RODRIGUEZ NARANJOAún no hay calificaciones
- Importancia Biologica de Las ProteinasDocumento9 páginasImportancia Biologica de Las Proteinasjuan angelAún no hay calificaciones
- Tarea 2Documento13 páginasTarea 2Karol MosqueraAún no hay calificaciones
- Actividades Tema de La CélulaDocumento7 páginasActividades Tema de La CélulamarinmolinaelenaAún no hay calificaciones
- Lectura en InglesDocumento6 páginasLectura en Inglesgabi salgueroAún no hay calificaciones
- Organelas MPDocumento4 páginasOrganelas MPPriscilaAún no hay calificaciones
- Taller 1 BioquímicaDocumento13 páginasTaller 1 BioquímicaSantiago VargasAún no hay calificaciones
- Cardona - Las Proteínas. de La Estructura Primaria A La Cuaternaria.Documento9 páginasCardona - Las Proteínas. de La Estructura Primaria A La Cuaternaria.perezsanchezsharonAún no hay calificaciones
- Preguntas de RepasoDocumento17 páginasPreguntas de RepasoBananero Ramos0% (1)
- Preguntas Estructura ProteinasDocumento6 páginasPreguntas Estructura ProteinasArianna Lizeth Gonzalez AlmanzaAún no hay calificaciones
- Semana 2 MFHDocumento9 páginasSemana 2 MFHEdimar Moya100% (1)
- Taller 8 CompletoDocumento12 páginasTaller 8 CompletoThania GonzlezAún no hay calificaciones
- Actividad 4 - BGyCDocumento7 páginasActividad 4 - BGyCRocío LallanaAún no hay calificaciones
- Dieta Post Cirugía ProctológicaDocumento3 páginasDieta Post Cirugía ProctológicaAndrés OsorioAún no hay calificaciones
- Psalud Informe Mensual de Atenciones Julio 2019 DR Sosa Honduras Fosforera-2Documento349 páginasPsalud Informe Mensual de Atenciones Julio 2019 DR Sosa Honduras Fosforera-2Geancarlo SosaAún no hay calificaciones
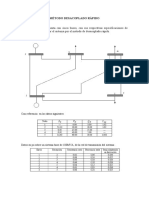
- Método Desacoplado RápidoDocumento8 páginasMétodo Desacoplado RápidoBeat FreAún no hay calificaciones
- Felix Carvajal Puebla Tarea 2Documento7 páginasFelix Carvajal Puebla Tarea 2Lavado DE Cobertores Donoso Pilar100% (1)
- Seminario 5 OctubreDocumento5 páginasSeminario 5 OctubredorzuohansAún no hay calificaciones
- Historia Del ArteDocumento8 páginasHistoria Del ArteGUIGIORGIAún no hay calificaciones
- Inventario Julio 2022Documento2 páginasInventario Julio 2022bethy rojas yujraAún no hay calificaciones
- El Hombre Como Ser Vicioso SocialDocumento6 páginasEl Hombre Como Ser Vicioso SocialAnthony RamirezAún no hay calificaciones
- Técnicas de Modelado GeométricoDocumento40 páginasTécnicas de Modelado GeométricoGabryela RcoAún no hay calificaciones
- 9.4 Reporte de ValorizacionDocumento15 páginas9.4 Reporte de ValorizacionWILDER JESUS GUEVARA MUÑOZAún no hay calificaciones
- Informe de Laboratorio Nº. 4 Física ElectrónicaDocumento15 páginasInforme de Laboratorio Nº. 4 Física ElectrónicaANGELITO240100% (1)
- Preparación de Núcleo de Roca Como Probeta Cilíndrica de Acuerdo Con La Norma Astm d4543Documento21 páginasPreparación de Núcleo de Roca Como Probeta Cilíndrica de Acuerdo Con La Norma Astm d4543Manuel Orellana PereiraAún no hay calificaciones
- Proyecto Tesis Con Las Intro Terminar Este!!!Documento73 páginasProyecto Tesis Con Las Intro Terminar Este!!!Sebastián AhumadaAún no hay calificaciones
- Antologia MilagrosDocumento5 páginasAntologia MilagrosRichard MillerAún no hay calificaciones
- Cuidados Partos - EnfermeriaDocumento3 páginasCuidados Partos - EnfermeriasandyvetasAún no hay calificaciones
- Grills Dentales PlantillaDocumento3 páginasGrills Dentales Plantillacrismana2006Aún no hay calificaciones
- Conversion Coordenadas Gps - UtmDocumento13 páginasConversion Coordenadas Gps - UtmMARY07abrAún no hay calificaciones
- Metodo de Deformaciones AngularesDocumento22 páginasMetodo de Deformaciones AngularesSaira J M Estela100% (1)
- Preguntas Quemados-1seminarioDocumento4 páginasPreguntas Quemados-1seminarioCarmen Salazar BahamondeAún no hay calificaciones
- Practica 3Documento13 páginasPractica 3Aby Chañi ArapaAún no hay calificaciones
- Calorimetría Práctica 7Documento5 páginasCalorimetría Práctica 7Rosalba HernándezAún no hay calificaciones
- 54716-01 Cme24 Bmax09-BslectDocumento28 páginas54716-01 Cme24 Bmax09-BslectjmmunozherreroAún no hay calificaciones
- Replica A Animal Polietico - Dra. Acevedo WhitehouseDocumento5 páginasReplica A Animal Polietico - Dra. Acevedo WhitehouseLucía FerreccioAún no hay calificaciones
- Dibujo Tecnico CruzDocumento12 páginasDibujo Tecnico CruzLeofeld ArriojaAún no hay calificaciones
- Apuntes de EtologíaDocumento80 páginasApuntes de EtologíaLuis Antonio Martínez FrancoAún no hay calificaciones
- Elaboracion de Chirizo ParrilleroDocumento8 páginasElaboracion de Chirizo ParrilleroAlvaro LedezmaAún no hay calificaciones