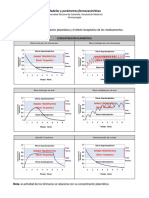
También podría gustarte
- Libro - Biofarmacia y Farmacocinetica Clinica PDFDocumento301 páginasLibro - Biofarmacia y Farmacocinetica Clinica PDFJosselyn Gutierrez100% (3)
- Parametros Farmacocineticos Biodisponibilidad y Volumen Aparente de DistribucionDocumento42 páginasParametros Farmacocineticos Biodisponibilidad y Volumen Aparente de DistribucionGise Hinostroza100% (1)
- Introducció A La BiofarmaciaDocumento74 páginasIntroducció A La Biofarmaciayohanvilk100% (1)
- Cuadro Comparativo de Las Vías de Administración de MedicamentosDocumento5 páginasCuadro Comparativo de Las Vías de Administración de MedicamentosDani100% (1)
- Estudios de BioequivalenciaDocumento12 páginasEstudios de BioequivalenciajuanAún no hay calificaciones
- Semana 1 Introduccion A La Biof-Fcocin. BF - FKDocumento30 páginasSemana 1 Introduccion A La Biof-Fcocin. BF - FKricardo100% (1)
- Interacciones FarmacológicasDocumento792 páginasInteracciones FarmacológicasGeovanny Leonardo Plaza Izurieta100% (1)
- Biodisponibilidad y BioequivalenciaDocumento28 páginasBiodisponibilidad y BioequivalenciaEddy CarbajalAún no hay calificaciones
- Biodisponibilidad y Bioequivalencia-Exposicion - Biofarmacia.Documento37 páginasBiodisponibilidad y Bioequivalencia-Exposicion - Biofarmacia.leisyAún no hay calificaciones
- Tema 7 BiodisponibilidadDocumento17 páginasTema 7 BiodisponibilidadCecilia Gladys Cohaila AcevedoAún no hay calificaciones
- Microbiota y microbiomaterapia en gastroenterología CMG 5De EverandMicrobiota y microbiomaterapia en gastroenterología CMG 5Aún no hay calificaciones
- Compatibilidad Medicamentos en YDocumento1 páginaCompatibilidad Medicamentos en YVicky Tina100% (2)
- Receta ImssDocumento1 páginaReceta Imssraul castillo gomezAún no hay calificaciones
- Biodisponibilidad y BioequivalenciaDocumento48 páginasBiodisponibilidad y BioequivalenciaRocio DelgadoAún no hay calificaciones
- Tema 23 Biodisponibilidad2 OCW PDFDocumento18 páginasTema 23 Biodisponibilidad2 OCW PDFsteve figueroaAún no hay calificaciones
- Grupo Nº7 Biodisponibilidad y Bioequivalencia (1) ...Documento29 páginasGrupo Nº7 Biodisponibilidad y Bioequivalencia (1) ...oscarAún no hay calificaciones
- Monocompartimental PDFDocumento57 páginasMonocompartimental PDFriesgos33100% (1)
- Analisis de Un Reporte de BioequivalenciaDocumento40 páginasAnalisis de Un Reporte de BioequivalenciaJesús Poot IslasAún no hay calificaciones
- Procesos BiofarmacéuticosDocumento8 páginasProcesos BiofarmacéuticosJhon Cabezas MoranAún no hay calificaciones
- MonocompartimentalDocumento56 páginasMonocompartimentalJeannie RouxAún no hay calificaciones
- AceitunoDocumento30 páginasAceitunoLu BlindAún no hay calificaciones
- Biodisponibilidad UltimoDocumento25 páginasBiodisponibilidad UltimoMargot Mendoza SalasAún no hay calificaciones
- Biodisponibilidad y BioequivalenciaDocumento15 páginasBiodisponibilidad y BioequivalenciaCarolina Herrera SuelperesAún no hay calificaciones
- BE FDAModulo2 4Documento172 páginasBE FDAModulo2 4angie valencia hurtadoAún no hay calificaciones
- BIOQUIVALENCIADocumento10 páginasBIOQUIVALENCIAIsabel CristinaAún no hay calificaciones
- Biodisponibilidad y BioequivalenciaDocumento40 páginasBiodisponibilidad y Bioequivalenciadoly adelidAún no hay calificaciones
- Sesión 14 de Febrero - Modelos y Parámetros FarmacocinéticosDocumento18 páginasSesión 14 de Febrero - Modelos y Parámetros FarmacocinéticosWilliam Nicolas Pozo NarvaezAún no hay calificaciones
- Trabajao InvestigacvionDocumento17 páginasTrabajao InvestigacvionMarcos Hurtado CalderonAún no hay calificaciones
- T09. Parámetros de La Biodisponibilidad - 2021Documento49 páginasT09. Parámetros de La Biodisponibilidad - 2021MILAGROS KATHERINE VILLENA GRADOSAún no hay calificaciones
- BIOEQUIVALENCIADocumento7 páginasBIOEQUIVALENCIAWesResAún no hay calificaciones
- BiodisponibilidadDocumento24 páginasBiodisponibilidadquintanilla_asesorAún no hay calificaciones
- Introduccion Farmacologia General Obstetricia 2014 (Clase 1)Documento59 páginasIntroduccion Farmacologia General Obstetricia 2014 (Clase 1)OscarAún no hay calificaciones
- Biodisponibilidad y Bioequivalencia 2023-2Documento92 páginasBiodisponibilidad y Bioequivalencia 2023-2andreaAún no hay calificaciones
- Programa Biofarmacia y Farmacocinética 2-2020-Pregrado DefinitivoDocumento4 páginasPrograma Biofarmacia y Farmacocinética 2-2020-Pregrado DefinitivoIsabella Blanco MedinaAún no hay calificaciones
- UNIDAD 1 FARMACOCINÉTICA Guia de Trabajos PracticosDocumento12 páginasUNIDAD 1 FARMACOCINÉTICA Guia de Trabajos Practicosfelipe islaAún no hay calificaciones
- ABC BioequiDocumento30 páginasABC BioequiAdriana Soto MijaresAún no hay calificaciones
- Biodisponibilidad y Bioequivalencia Exposicion BiofarmaciaDocumento35 páginasBiodisponibilidad y Bioequivalencia Exposicion BiofarmaciaAaron Garcia SanchezAún no hay calificaciones
- Monitoreo Clínico PK-PD FM-UNAMDocumento35 páginasMonitoreo Clínico PK-PD FM-UNAMJoguard LofcraftAún no hay calificaciones
- Trabajo Biodisponibilidad - BioequivalenciaDocumento7 páginasTrabajo Biodisponibilidad - BioequivalenciaAmy Lissbet Abad CarreñoAún no hay calificaciones
- Reporte BioequivalenciaDocumento17 páginasReporte BioequivalenciaDany GarcesAún no hay calificaciones
- Práctica 10 - Estándares de Bioequivalencia Aplicación Del BCS - Sistema de Clasificación BiofarmacéutDocumento11 páginasPráctica 10 - Estándares de Bioequivalencia Aplicación Del BCS - Sistema de Clasificación BiofarmacéutEro CoylaAún no hay calificaciones
- Biodisponibilidad y BioequivalenciaDocumento8 páginasBiodisponibilidad y BioequivalenciaWesResAún no hay calificaciones
- Biodisponibilidad y Bioequivalencia.Documento28 páginasBiodisponibilidad y Bioequivalencia.Anel Jareth Peña IndaAún no hay calificaciones
- Farmacocinética Clínica Aspectos Teórico-Prácticos y Su Aplicación en El Laboratorio Clínico Clase 2 09-04-2022Documento28 páginasFarmacocinética Clínica Aspectos Teórico-Prácticos y Su Aplicación en El Laboratorio Clínico Clase 2 09-04-2022alejandra soledad alvarado neiraAún no hay calificaciones
- Biodisponibilidad y Bioequivalencia+MakDocumento41 páginasBiodisponibilidad y Bioequivalencia+MakJessica Benavides BermudezAún no hay calificaciones
- Biodisponibilidad de FármacosDocumento50 páginasBiodisponibilidad de Fármacosmanuel benitezAún no hay calificaciones
- BiofarmaciaDocumento4 páginasBiofarmaciaEstefania VelascoAún no hay calificaciones
- 1-Introducció A La BiofarmaciaDocumento77 páginas1-Introducció A La BiofarmaciaMarcos Antonio TaurusAún no hay calificaciones
- Silabo de BIOFARMACIA Y FARMACOCINETICADocumento10 páginasSilabo de BIOFARMACIA Y FARMACOCINETICAPabloCoilaMirandaAún no hay calificaciones
- Patron Hito 2Documento5 páginasPatron Hito 2MARIA FERNANDA RIVERA RAMIREZAún no hay calificaciones
- Correlacion Invivo InvitroDocumento8 páginasCorrelacion Invivo InvitroCieloAún no hay calificaciones
- Articulo de BiodispobilidadDocumento11 páginasArticulo de BiodispobilidadJulio SantillanAún no hay calificaciones
- Temas 2 y 3. N Consideraciones BiofarmacéuticasDocumento27 páginasTemas 2 y 3. N Consideraciones Biofarmacéuticassimply_white408267% (3)
- Clase 7 Biofarmacia y BioequivalenciaDocumento27 páginasClase 7 Biofarmacia y BioequivalenciaJoubert Diaz MarinAún no hay calificaciones
- Evaluación Eficacia y Estabilidad de MedicamentosDocumento55 páginasEvaluación Eficacia y Estabilidad de MedicamentosKathy CerdaAún no hay calificaciones
- Biodisponibilidad y Su Proceso FarmacológicoDocumento6 páginasBiodisponibilidad y Su Proceso FarmacológicoCathy Díaz PérezAún no hay calificaciones
- Biofarmacia Tarea #1Documento3 páginasBiofarmacia Tarea #1roberth panchanaAún no hay calificaciones
- Guia para Establecer Las Bases de La Realización de Estudios de Biodisponibilidad y Bioequivalencia de Productos Farmacéuticos de Administración OralDocumento6 páginasGuia para Establecer Las Bases de La Realización de Estudios de Biodisponibilidad y Bioequivalencia de Productos Farmacéuticos de Administración OralGilberto Perez MartinezAún no hay calificaciones
- Biodisponibilidad y BioequivalenciaDocumento16 páginasBiodisponibilidad y BioequivalenciaManuel Rios GonzalesAún no hay calificaciones
- Taller 9Documento3 páginasTaller 9Angélica MorenoAún no hay calificaciones
- Diseños Experimentales en BioequivalenciaDocumento12 páginasDiseños Experimentales en BioequivalenciaMaría Elena GaminoAún no hay calificaciones
- Principales procedimientos quirúrgicos CMG 6De EverandPrincipales procedimientos quirúrgicos CMG 6Aún no hay calificaciones
- Ciencia regulatoria: Medicamentos bio y su relevancia para la saludDe EverandCiencia regulatoria: Medicamentos bio y su relevancia para la saludAún no hay calificaciones
- Diapositivas Enfermeria BasicaDocumento11 páginasDiapositivas Enfermeria BasicaEstefany YanezAún no hay calificaciones
- Relajantes NeuromuscularesDocumento9 páginasRelajantes NeuromuscularesCris RamirezAún no hay calificaciones
- Articulo 5Documento4 páginasArticulo 5Juan SolanoAún no hay calificaciones
- Dilución y Estabilidad de AntimicrobianosDocumento3 páginasDilución y Estabilidad de AntimicrobianosFloOr CastroAún no hay calificaciones
- BenzodiacepinasDocumento5 páginasBenzodiacepinasIsa Vasquez PAún no hay calificaciones
- CalcioantagonistasDocumento28 páginasCalcioantagonistasCITLALI YATZIL SANDOVAL CATALANAún no hay calificaciones
- Med PediatriaDocumento6 páginasMed PediatriaAngel HCAún no hay calificaciones
- Farmacos de OdontologiaDocumento23 páginasFarmacos de Odontologiayesenia gallegosAún no hay calificaciones
- Presentación 29 MayoDocumento14 páginasPresentación 29 MayoEdgar Pérez IUAún no hay calificaciones
- Elementos de Una Receta MédicaDocumento1 páginaElementos de Una Receta MédicaKevin Maruri100% (1)
- Estadistica Drogas TlalnepantlaDocumento11 páginasEstadistica Drogas TlalnepantlaKaren RamírezAún no hay calificaciones
- Farmacología FarmacocinéticaDocumento111 páginasFarmacología FarmacocinéticaSofíaAún no hay calificaciones
- Nucleo Almdes Mednom Codigo - Pre Codigo - MED: Medf F Medc NC Medp RESDocumento2120 páginasNucleo Almdes Mednom Codigo - Pre Codigo - MED: Medf F Medc NC Medp RESJose Luis Pallarco PaucarAún no hay calificaciones
- Examen ExcelDocumento618 páginasExamen Excelsandrapar.cortesAún no hay calificaciones
- Esomeprazol 20 MG 40 MGDocumento15 páginasEsomeprazol 20 MG 40 MGClare BaliAún no hay calificaciones
- Axel 22.08.23Documento1 páginaAxel 22.08.23Alberto LpzAún no hay calificaciones
- Petitorio de Medicamentos y BiomédicosDocumento61 páginasPetitorio de Medicamentos y Biomédicos6gyqfzbvyvAún no hay calificaciones
- EstazolamDocumento3 páginasEstazolamvalentinoAún no hay calificaciones
- Arsenal Farmacológico HBV 2020-2021Documento58 páginasArsenal Farmacológico HBV 2020-2021internos medicinaAún no hay calificaciones
- Principales Farmacos para Infusion IntravenosaDocumento4 páginasPrincipales Farmacos para Infusion IntravenosaEdwardo BrownAún no hay calificaciones
- Liberación y AbsorciónDocumento9 páginasLiberación y AbsorciónBicho Garcia DuranAún no hay calificaciones
- Asi ArgentinaDocumento11 páginasAsi ArgentinaAndreaAún no hay calificaciones
- Flashcards MedicamentosDocumento7 páginasFlashcards MedicamentosAna DoloresAún no hay calificaciones
- Medicamentos Nula RotacionDocumento30 páginasMedicamentos Nula RotacionCLAUDIA MARTINEZAún no hay calificaciones
- Profilaxis AntifungicaDocumento24 páginasProfilaxis AntifungicajoelAún no hay calificaciones
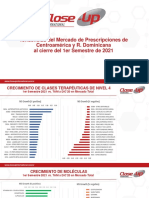
- Tendencia de Prescripciones Centroamerica y R Dominicana 1632189924Documento9 páginasTendencia de Prescripciones Centroamerica y R Dominicana 1632189924waleskaAún no hay calificaciones