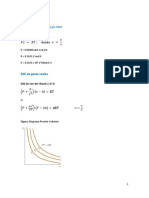
También podría gustarte
- 10ma SemanaDocumento127 páginas10ma SemanaAldahir BernalAún no hay calificaciones
- 10ma SemanaDocumento127 páginas10ma SemanaAldahir BernalAún no hay calificaciones
- Comprobaciones ELU (Barra) - en - Punto - Mas - DesfavorableDocumento7 páginasComprobaciones ELU (Barra) - en - Punto - Mas - DesfavorableLexAún no hay calificaciones
- Dinamica EjerciciosDocumento8 páginasDinamica EjerciciosKeyla LupucheAún no hay calificaciones
- 0 0 Completo-3Documento10 páginas0 0 Completo-3Leonel PeñaAún no hay calificaciones
- Vaciado de Tanques Cono Truncado y EsfericoDocumento4 páginasVaciado de Tanques Cono Truncado y Esferico1957noraperezAún no hay calificaciones
- Determinación del coeficiente de Poisson del aireDocumento12 páginasDeterminación del coeficiente de Poisson del aireCristhian VargasAún no hay calificaciones
- Poisson HenryyDocumento11 páginasPoisson HenryyNils Cristian RodriguezAún no hay calificaciones
- Retardo en La Conmutacion de Un Convertidor Ac DC Trifasico Tipo PuenteDocumento5 páginasRetardo en La Conmutacion de Un Convertidor Ac DC Trifasico Tipo PuenteCarolina BastidasAún no hay calificaciones
- Guia DinamicaDocumento6 páginasGuia DinamicaHernán Felipe Pacheco HuichalafAún no hay calificaciones
- Post3 LControlDocumento11 páginasPost3 LControlmariaAún no hay calificaciones
- Termodinámica EcuacionesDocumento33 páginasTermodinámica EcuacionesDiego VHAún no hay calificaciones
- Trabajo Autonomo 5 - 2P - Ericka Llangari 8-1 PDFDocumento7 páginasTrabajo Autonomo 5 - 2P - Ericka Llangari 8-1 PDFGaby ArmijosAún no hay calificaciones
- Cuestionario Previo 4Documento15 páginasCuestionario Previo 4Jose Antonio VazquezAún no hay calificaciones
- Practica 03, VIBRACIONES LIBRES DE SISTEMAS DE 1GLDocumento5 páginasPractica 03, VIBRACIONES LIBRES DE SISTEMAS DE 1GLCarlos HernándezAún no hay calificaciones
- Unidad No.9Documento5 páginasUnidad No.9Melissa MejiaAún no hay calificaciones
- Unidad No.9Documento5 páginasUnidad No.9Melissa MejiaAún no hay calificaciones
- Taller Modelamiento Tanques Cascada1Documento6 páginasTaller Modelamiento Tanques Cascada1JUAN CAMILO FERREIRA RODRIGUEZAún no hay calificaciones
- Aplicaciones A Gases RealesDocumento38 páginasAplicaciones A Gases RealesAldoCabreraFernandezAún no hay calificaciones
- 2 T Inv de Laplace - Uac-2Documento24 páginas2 T Inv de Laplace - Uac-2Gonzalo Sanchez CarmonaAún no hay calificaciones
- Determinación Cp/Cv aireDocumento6 páginasDeterminación Cp/Cv aireAaron GuzmanAún no hay calificaciones
- P5.2.1-UPM Series de FourierDocumento26 páginasP5.2.1-UPM Series de FouriercelsoAún no hay calificaciones
- Solucionario: Universidad Nacional de IngenieriaDocumento4 páginasSolucionario: Universidad Nacional de IngenieriaAlvaro RodrigoAún no hay calificaciones
- 1 Parcial SolucionarioDocumento3 páginas1 Parcial SolucionarioGlendaAún no hay calificaciones
- 1 Parcial SolucionarioDocumento3 páginas1 Parcial SolucionarioRogher Uluri YanaAún no hay calificaciones
- Serie 3 - ResoluciónDocumento26 páginasSerie 3 - Resolucióneliana vegaAún no hay calificaciones
- Guía Laboratorio 1Documento5 páginasGuía Laboratorio 1AntoniaAún no hay calificaciones
- Análisis del ciclo Otto con CyclepadDocumento8 páginasAnálisis del ciclo Otto con CyclepadJuan GámezAún no hay calificaciones
- EJERCICIOS DE CANALES. Mecánica de FluidosDocumento5 páginasEJERCICIOS DE CANALES. Mecánica de FluidosJhoe LozaAún no hay calificaciones
- 2B 3Documento6 páginas2B 3christianCAMOAún no hay calificaciones
- Ejercicios de práctica de laboratorio de fisicoquímica sobre procesos termodinámicosDocumento6 páginasEjercicios de práctica de laboratorio de fisicoquímica sobre procesos termodinámicosJose Gomez Alvarez50% (2)
- Transporte y Almacenamiento de HidrocarburosDocumento13 páginasTransporte y Almacenamiento de HidrocarburosrodrigoAún no hay calificaciones
- TEORÍA DE TUBERÍAS EN SERIE Y EN PARALELODocumento14 páginasTEORÍA DE TUBERÍAS EN SERIE Y EN PARALELOJoseph VasquezAún no hay calificaciones
- IIIIIIIIDocumento16 páginasIIIIIIIIHaidee Cusi AceroAún no hay calificaciones
- Circulo de MohrDocumento36 páginasCirculo de MohrJunior MonteroAún no hay calificaciones
- Funcion SenoidalDocumento7 páginasFuncion SenoidalAndrea SerranoAún no hay calificaciones
- Solucionario 2do Parcial Fis102 Ver2022Documento5 páginasSolucionario 2do Parcial Fis102 Ver2022Camila Zapata AriasAún no hay calificaciones
- Problemas Series Fourier Tarea - En.esDocumento3 páginasProblemas Series Fourier Tarea - En.esDumas DumasAún no hay calificaciones
- Absorcion - Guia de Tablas y Correlaciones Ver.4Documento9 páginasAbsorcion - Guia de Tablas y Correlaciones Ver.4TobiasAún no hay calificaciones
- Ecuación de ClapeyronDocumento9 páginasEcuación de ClapeyronEdward AlexanderAún no hay calificaciones
- Sistemas dinámicos de primer orden: análisis de procesosDocumento36 páginasSistemas dinámicos de primer orden: análisis de procesosSebastian Jose Diaz VillarrealAún no hay calificaciones
- Los Chicos de Químicas Act 5Documento17 páginasLos Chicos de Químicas Act 5fonsAún no hay calificaciones
- Algoritmo Banco de BombasDocumento5 páginasAlgoritmo Banco de BombasAngela CalatayudAún no hay calificaciones
- 2 5 PDFDocumento3 páginas2 5 PDFJuan ManuAún no hay calificaciones
- Actividad 9 FinalDocumento11 páginasActividad 9 FinalLuis Fernando ZambranoAún no hay calificaciones
- Ejercicio Matlab Semi Lotes FinalDocumento12 páginasEjercicio Matlab Semi Lotes FinalWendy Johana RengifoAún no hay calificaciones
- Clase 2.3. EdE - SeminarioDocumento10 páginasClase 2.3. EdE - SeminarioVale IturraAún no hay calificaciones
- Problemas de Aplicación de Reactores Ideales - Garcia, Rique, Sanchez, Taboada & UrbanoDocumento18 páginasProblemas de Aplicación de Reactores Ideales - Garcia, Rique, Sanchez, Taboada & UrbanoTABOADA ESTRADA DANIELA GIMENAAún no hay calificaciones
- Examen RA C Ord 20 21Documento3 páginasExamen RA C Ord 20 21antonio1575Aún no hay calificaciones
- Trabajo Completo Control Primera FaseDocumento66 páginasTrabajo Completo Control Primera FaseJuniior Cabrera BolivarAún no hay calificaciones
- Ecuación de la energía en canales abiertosDocumento21 páginasEcuación de la energía en canales abiertosMariaelena HuamaniAún no hay calificaciones
- CUESTIONARIO HidraulicaDocumento16 páginasCUESTIONARIO HidraulicaDaniela Castro100% (1)
- P1 - Control AUt.Documento14 páginasP1 - Control AUt.Melisa ParedesAún no hay calificaciones
- Clase 18-11, 1Documento6 páginasClase 18-11, 1Marco Antonio Campos VeraAún no hay calificaciones
- Ecuaciones de Energía Toro 2Documento16 páginasEcuaciones de Energía Toro 2AlejandroAún no hay calificaciones
- Ejercicios FT, Transformada de Laplace y Linealización PDFDocumento5 páginasEjercicios FT, Transformada de Laplace y Linealización PDFPedro Luis BohorquezAún no hay calificaciones
- Informe Dittus BoelterDocumento15 páginasInforme Dittus BoelterElena Ferrer MartinezAún no hay calificaciones
- Intercambiadores de CalorDocumento18 páginasIntercambiadores de Calorisabella quijano polaniaAún no hay calificaciones
- Transferencia de calor en régimen transitorioDocumento16 páginasTransferencia de calor en régimen transitorioisraelAún no hay calificaciones
- Funcion de TransferenciaDocumento9 páginasFuncion de TransferenciaBrandy Tacca HuamanAún no hay calificaciones
- Práctica Calificada 2 - Sistema de Control ModernoDocumento17 páginasPráctica Calificada 2 - Sistema de Control ModernoPedro SuarezAún no hay calificaciones
- Ejercicios de Integrales y Ecuaciones Integrales-DiferencialesDe EverandEjercicios de Integrales y Ecuaciones Integrales-DiferencialesAún no hay calificaciones
- Relaciones TermodinámicasDocumento22 páginasRelaciones TermodinámicasJoaquín GarcíaAún no hay calificaciones
- Termodinámica I: Fugacidad y coeficiente de fugacidad de gases purosDocumento16 páginasTermodinámica I: Fugacidad y coeficiente de fugacidad de gases purosJoaquín GarcíaAún no hay calificaciones
- Ciclos TermicosDocumento43 páginasCiclos TermicosLesly PerezAún no hay calificaciones
- Termo 2 24-08Documento5 páginasTermo 2 24-08Joaquín GarcíaAún no hay calificaciones
- Equilibrio Liquido Vapor Informe 1Documento11 páginasEquilibrio Liquido Vapor Informe 1Joaquín GarcíaAún no hay calificaciones
- 12va SemanaDocumento25 páginas12va SemanaJoaquín GarcíaAún no hay calificaciones
- Laboratorio de Alimentos-Practica Nº1Documento10 páginasLaboratorio de Alimentos-Practica Nº1Joaquín GarcíaAún no hay calificaciones
- SEMANA #1 IntroducciónDocumento24 páginasSEMANA #1 IntroducciónJoaquín GarcíaAún no hay calificaciones
- Valoraciones de precipitación: Fundamentos y métodos de Mohr y VolhardDocumento34 páginasValoraciones de precipitación: Fundamentos y métodos de Mohr y VolhardJoaquín GarcíaAún no hay calificaciones
- Reología en Fluidos de PerforacionDocumento24 páginasReología en Fluidos de PerforacionFranz Kevin Luna AguilarAún no hay calificaciones
- Práctica 4 Campo Electrico Reporte XFDocumento9 páginasPráctica 4 Campo Electrico Reporte XFJulian Gerardo Quispe MallquiAún no hay calificaciones
- CORAZADocumento3 páginasCORAZAVanessaAún no hay calificaciones
- Fisica 2 YjhgDocumento96 páginasFisica 2 YjhgMiranda Castillo100% (1)
- 3ra Práctica Calificada Ee210n, Ciclo 2020-1Documento2 páginas3ra Práctica Calificada Ee210n, Ciclo 2020-1Diego Salazar UrbinaAún no hay calificaciones
- Taller en Clase Ley de HookeDocumento5 páginasTaller en Clase Ley de HookeTatiana RojasAún no hay calificaciones
- Pendulo DobleDocumento16 páginasPendulo DobleOrlando Perez CarrilloAún no hay calificaciones
- TIF FENOMENOS MARTES 2do AvanceDocumento18 páginasTIF FENOMENOS MARTES 2do AvanceAnthonio Casos MendozaAún no hay calificaciones
- Mruv Pre-U 14-03-2023Documento3 páginasMruv Pre-U 14-03-2023Alonso PumaAún no hay calificaciones
- Eva Parcial Mec de Mat 1 2021 10Documento5 páginasEva Parcial Mec de Mat 1 2021 10Beltran GutierrezAún no hay calificaciones
- Ejercicio Prctico 1 - Mezcla Rpida y LentaDocumento2 páginasEjercicio Prctico 1 - Mezcla Rpida y LentaLeyda TejedorAún no hay calificaciones
- Taller Completo Parte A, B, C Balance de EnergiaDocumento10 páginasTaller Completo Parte A, B, C Balance de EnergiaDiana Caroline Arevalo VelaAún no hay calificaciones
- Practica 5Documento5 páginasPractica 5EDISSON ANDRES VILLA AVILAAún no hay calificaciones
- Mecánica de Fluidos II: Semana 6 PracticaDocumento13 páginasMecánica de Fluidos II: Semana 6 PracticaLuis ArosteguiAún no hay calificaciones
- Reticula Do 1Documento5 páginasReticula Do 1Carlos ParedesAún no hay calificaciones
- Maquina Ster MicasDocumento59 páginasMaquina Ster MicasPAOLA ANDREA QUENALLATA CHIPANAAún no hay calificaciones
- Ensayo de Tracción en MetalesDocumento13 páginasEnsayo de Tracción en MetalesJohn Diego GutierrezAún no hay calificaciones
- Circuitos Trifásicos LinealesDocumento9 páginasCircuitos Trifásicos LinealesluciaAún no hay calificaciones
- Actividad #07. - Magnetismo Fuerza MagnéticaDocumento5 páginasActividad #07. - Magnetismo Fuerza MagnéticaMelany Geraldine PecheAún no hay calificaciones
- Ipfl Lab N°2 Grupo 3Documento6 páginasIpfl Lab N°2 Grupo 3Jhon Brandon Rojas YauriAún no hay calificaciones
- Practica No.9 Segunda Ley de NewtonDocumento8 páginasPractica No.9 Segunda Ley de NewtonPérez Hernández AlejandroAún no hay calificaciones
- Practica 1 Primer Parcial 1 - 2019Documento2 páginasPractica 1 Primer Parcial 1 - 2019Bicmar José Carreón RejasAún no hay calificaciones
- Fatiga PNDDocumento65 páginasFatiga PNDYamiBakuraT3M0Aún no hay calificaciones
- Transmision de Calor en Un Tanque AgitadoDocumento12 páginasTransmision de Calor en Un Tanque AgitadoLizet Tincuta100% (1)
- TipologiasDocumento19 páginasTipologiasMora MurielAún no hay calificaciones
- Tema - 01 - Fisica Del SonidoDocumento18 páginasTema - 01 - Fisica Del SonidoDustin SantanderAún no hay calificaciones
- Dinamica 1 - UPC PDFDocumento45 páginasDinamica 1 - UPC PDFCristhian CarlosAún no hay calificaciones