También podría gustarte
- Bioenergética. Introducción a la teoría quimiosmóticaDe EverandBioenergética. Introducción a la teoría quimiosmóticaAún no hay calificaciones
- PP UNIDAD 5 - ANTIBIÓTICOS 2021-PenicilinaDocumento37 páginasPP UNIDAD 5 - ANTIBIÓTICOS 2021-PenicilinaFernanda TorresAún no hay calificaciones
- Cap. 43 y 44. B Lactámicos, Tetraciclinas, Macrólidos y ClindamicinaDocumento22 páginasCap. 43 y 44. B Lactámicos, Tetraciclinas, Macrólidos y ClindamicinabetsaidanaranjoAún no hay calificaciones
- Antibioticos BetalactamicosDocumento10 páginasAntibioticos BetalactamicosSilvyAún no hay calificaciones
- Acceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017De EverandAcceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017Aún no hay calificaciones
- Antibioticos de Espectro ReducidoDocumento16 páginasAntibioticos de Espectro ReducidoRuth Nancy LavillaAún no hay calificaciones
- 1 BetalactámicosDocumento22 páginas1 Betalactámicosdanielamedicina170% (1)
- Informe 2Documento4 páginasInforme 2Peter HuamanAún no hay calificaciones
- Nori Janeth Juzga OrdoñezDocumento3 páginasNori Janeth Juzga OrdoñezScopy PrinterAún no hay calificaciones
- Folleto Penicilina-Quimica OrganicaDocumento2 páginasFolleto Penicilina-Quimica OrganicaMartin Nicolas Campos Riveros100% (1)
- TP 6 - Inhibidores de ParedDocumento46 páginasTP 6 - Inhibidores de ParedALISON ANDREA BERROCAL SALAZARAún no hay calificaciones
- S7amif Antibioticos Que Actuan Sobre La Envoltura Celular Bacteriana Desde Pag 9Documento22 páginasS7amif Antibioticos Que Actuan Sobre La Envoltura Celular Bacteriana Desde Pag 9Maryory ChAún no hay calificaciones
- Tema 8 - Mecanismos de Acción y Resistencia A Los AntibióticosDocumento21 páginasTema 8 - Mecanismos de Acción y Resistencia A Los Antibióticosconnie muñozAún no hay calificaciones
- Farmacoquímica - Sesión 3Documento38 páginasFarmacoquímica - Sesión 3anhyelyAún no hay calificaciones
- Betalactamicos GeneralidadesDocumento23 páginasBetalactamicos GeneralidadeszulimarAún no hay calificaciones
- Uwiener Farmacoquimica B Lactamicos 6-2019Documento46 páginasUwiener Farmacoquimica B Lactamicos 6-2019yessy Pujaico PalaciosAún no hay calificaciones
- Atb y Quimioterapicos I III 17Documento168 páginasAtb y Quimioterapicos I III 17Jorge MuroAún no hay calificaciones
- PDF 2 Español Transporte de LipidosDocumento15 páginasPDF 2 Español Transporte de LipidosMilena PulidoAún no hay calificaciones
- Encuesta Y ResumenDocumento15 páginasEncuesta Y ResumenLaura TorresAún no hay calificaciones
- TEORIA10 - CefalosporinasDocumento18 páginasTEORIA10 - CefalosporinasGriselFloresZavaletaAún no hay calificaciones
- Atb y Quimioterapicos IDocumento79 páginasAtb y Quimioterapicos ILeslie Rosado MerinoAún no hay calificaciones
- Articulo CientificoDocumento8 páginasArticulo CientificoTATIANA URBINAAún no hay calificaciones
- Farmacologia - Antibióticos Betalactamicos (I y II)Documento152 páginasFarmacologia - Antibióticos Betalactamicos (I y II)Fabricio Moncada SaavedraAún no hay calificaciones
- Practica 2 Ftyii - 1-2021.ucDocumento5 páginasPractica 2 Ftyii - 1-2021.uckevin caero100% (1)
- Capítulo 5 TayDocumento5 páginasCapítulo 5 TayKeny Glez GarzaAún no hay calificaciones
- Inhibidores de La Síntesis de Pared de MembranaDocumento4 páginasInhibidores de La Síntesis de Pared de MembranaBrando Mejia ZepedaAún no hay calificaciones
- 6 ANTIMICROBIANOS Dra Poma VDocumento53 páginas6 ANTIMICROBIANOS Dra Poma VAkiro PulidoAún no hay calificaciones
- Síntesis de CefalosporinasDocumento13 páginasSíntesis de CefalosporinasAngela NuñezAún no hay calificaciones
- Antibióticos BetalactámicosDocumento22 páginasAntibióticos BetalactámicosRakel Vasquez MaytaAún no hay calificaciones
- Antibioticos BetalactamicosDocumento7 páginasAntibioticos BetalactamicosBrigitte MaldonadoAún no hay calificaciones
- Unidad I: Estudio de La Vida y Su Base Química Procariotas. Célula BacterianaDocumento21 páginasUnidad I: Estudio de La Vida y Su Base Química Procariotas. Célula BacterianaValentina hinostroza AlvaradoAún no hay calificaciones
- Antimicrobianos IDocumento7 páginasAntimicrobianos IJimena Sanchez ZeaAún no hay calificaciones
- S0213005X03728730Documento14 páginasS0213005X03728730jimenzguzmanaAún no hay calificaciones
- Guia - Betalactamicos - PenicilinasDocumento9 páginasGuia - Betalactamicos - PenicilinasCátedra de Farmacología de la Escuela de Medicina de La UNIVERSIDAD DEL ZULIAAún no hay calificaciones
- Monografia de CefalexinaDocumento24 páginasMonografia de CefalexinaVioletta Castro CastilloAún no hay calificaciones
- AntibioticosDocumento18 páginasAntibioticosfernandabsotovAún no hay calificaciones
- Compuestos Antimicrobianos: Penicilinas NaturalesDocumento27 páginasCompuestos Antimicrobianos: Penicilinas NaturalesFelipe PradinesAún no hay calificaciones
- AntimicrobianosDocumento16 páginasAntimicrobianosFuukachi 7u7Aún no hay calificaciones
- BetalactamicosDocumento53 páginasBetalactamicoschwbaccaAún no hay calificaciones
- Mecanismo de Accion de Los AntibioticosDocumento53 páginasMecanismo de Accion de Los AntibioticosAlejandra Tenorio HernándezAún no hay calificaciones
- PENICILINASDocumento18 páginasPENICILINASGabbo BPAún no hay calificaciones
- Dokumen - Tips Faramacologia-Odontologiapdf PDFDocumento79 páginasDokumen - Tips Faramacologia-Odontologiapdf PDFyuvitzaAún no hay calificaciones
- Clase 11 FarmacoquimicaDocumento23 páginasClase 11 FarmacoquimicaPedro Alejandro Mesias SanchezAún no hay calificaciones
- LactamasDocumento20 páginasLactamasmunozdelgadokarlaAún no hay calificaciones
- 2021 Segunda Clase de Antibioticos y ClasificacionDocumento76 páginas2021 Segunda Clase de Antibioticos y Clasificacioningrid jamiAún no hay calificaciones
- Betalactamicos GlucopeptidosDocumento35 páginasBetalactamicos Glucopeptidosivan dugarte dugarteAún no hay calificaciones
- Clasificación de Los AntimicrobianosDocumento9 páginasClasificación de Los AntimicrobianosMario MegaduckAún no hay calificaciones
- 2da Clase - Penicilina y BetalactamicosDocumento58 páginas2da Clase - Penicilina y BetalactamicosLisania Millano100% (5)
- PENICILINADocumento3 páginasPENICILINAKHALIL EUGENIO ALI AVALOSAún no hay calificaciones
- Vademecum MayoDocumento63 páginasVademecum MayoSo VAún no hay calificaciones
- Antibiotic OsDocumento25 páginasAntibiotic OsDiana D DiosAún no hay calificaciones
- ATB 3. Betalactamicos 2022.versión FinalDocumento31 páginasATB 3. Betalactamicos 2022.versión FinalELEGMARAún no hay calificaciones
- Penicilinas - LecturioDocumento18 páginasPenicilinas - LecturioMARIA ALEXANDRA CARAZAS ZAPATAAún no hay calificaciones
- Caso Clinico 2Documento4 páginasCaso Clinico 2yuniorAún no hay calificaciones
- B LactamicosDocumento4 páginasB LactamicosLiseth SC100% (1)
- Inhibidores de La Pared (Autoguardado)Documento62 páginasInhibidores de La Pared (Autoguardado)Fer HernándezAún no hay calificaciones
- 2019 II Busqueda de Nuevos Farmacos (6287)Documento21 páginas2019 II Busqueda de Nuevos Farmacos (6287)Shierlley SantiAún no hay calificaciones
- Cuestionario BetalactamicosDocumento2 páginasCuestionario BetalactamicosRonald Ccoropuna CutimboAún no hay calificaciones
- GLUCOGENOGENESISDocumento2 páginasGLUCOGENOGENESISMARGARITA MOO CHANAún no hay calificaciones
- Semana 12 - Teoria QuimicaDocumento19 páginasSemana 12 - Teoria QuimicaLuciana AronesAún no hay calificaciones
- Practica de Botanica 2Documento7 páginasPractica de Botanica 2Howler25Aún no hay calificaciones
- Metabolismo de Los Carbohidratos DiapositivasDocumento6 páginasMetabolismo de Los Carbohidratos DiapositivasplimenAún no hay calificaciones
- Taller de Naturales DanielDocumento2 páginasTaller de Naturales DanielLuz Ramirez FigueroaAún no hay calificaciones
- Quimica Clinica-TripsinaDocumento4 páginasQuimica Clinica-TripsinaQUIMICO CLINICO WILLIANS SANCHEZ100% (1)
- GAMETOGÉNESISDocumento14 páginasGAMETOGÉNESISAshley Benites TuestasAún no hay calificaciones
- Anomalías en El Metabolismo de Los Lípidos.Documento4 páginasAnomalías en El Metabolismo de Los Lípidos.Emny EcAún no hay calificaciones
- Evaluacion Nucleo Celular Genetica MolecularDocumento2 páginasEvaluacion Nucleo Celular Genetica Molecularluisany camposAún no hay calificaciones
- CLASE #1 Presentacion, Toxicologia LaboralDocumento35 páginasCLASE #1 Presentacion, Toxicologia LaboralAriel PiñaAún no hay calificaciones
- Manual Nutricion Animal 6toDocumento125 páginasManual Nutricion Animal 6toMatias MuñozAún no hay calificaciones
- Respiracion de La PLanta de CañaDocumento18 páginasRespiracion de La PLanta de CañaJuan Carlos Telchi AbuawadAún no hay calificaciones
- Sistema Renina Angiotensina AldosteronaDocumento21 páginasSistema Renina Angiotensina AldosteronaHeidi Paola100% (2)
- Musculo Estriado EsqueléticoDocumento1 páginaMusculo Estriado EsqueléticoLibni Daniela HernándezAún no hay calificaciones
- Capítulo 7 - Enzimas - Mecanismo de AcciónDocumento23 páginasCapítulo 7 - Enzimas - Mecanismo de AcciónHéctor RosalesAún no hay calificaciones
- EnzimasDocumento5 páginasEnzimasAdrian LeónAún no hay calificaciones
- Fisio 1.contraccionDocumento32 páginasFisio 1.contraccionAlejandraAún no hay calificaciones
- AVU 2023 - Niveles de Organización - La Célula - Dra GambaDocumento44 páginasAVU 2023 - Niveles de Organización - La Célula - Dra GambaSabina Maribel GomezAún no hay calificaciones
- Alimentos - Bromatología 2Documento4 páginasAlimentos - Bromatología 2jean pierreAún no hay calificaciones
- La Recombinación Del ADNDocumento7 páginasLa Recombinación Del ADNYahir AriasAún no hay calificaciones
- Pregunta 1 y Marco SeminarioDocumento5 páginasPregunta 1 y Marco SeminarioCarlosSonoAún no hay calificaciones
- Alimentos en ServicioDocumento45 páginasAlimentos en ServicioClaren CingolaniAún no hay calificaciones
- Cuáles Son Los Principales Pigmentos Que Se Encuentran Dentro Del CloroplastoDocumento4 páginasCuáles Son Los Principales Pigmentos Que Se Encuentran Dentro Del CloroplastoGothardo Casanova TorresAún no hay calificaciones
- Plan de Desarrollo Curricular 2021Documento6 páginasPlan de Desarrollo Curricular 2021maruja olvea86% (7)
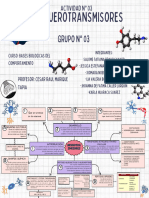
- NEUROTRASMISORESDocumento2 páginasNEUROTRASMISORESsalomebravosayagoAún no hay calificaciones
- Trabajo 3 Anatomia Sistema Digestivo.Documento7 páginasTrabajo 3 Anatomia Sistema Digestivo.facturamadigitalAún no hay calificaciones
- 2Documento3 páginas2María KarinaAún no hay calificaciones
- Diabetes MellitusDocumento51 páginasDiabetes Mellitusana100% (1)
- Examen Diagnostico BiologiaDocumento4 páginasExamen Diagnostico BiologiaNarda BernalAún no hay calificaciones
- Triptico de AntropologiaDocumento2 páginasTriptico de AntropologiaMarco Villafane EspinozaAún no hay calificaciones
- La revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaDe EverandLa revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaCalificación: 5 de 5 estrellas5/5 (202)
- Homo antecessor: El nacimiento de una especieDe EverandHomo antecessor: El nacimiento de una especieCalificación: 5 de 5 estrellas5/5 (1)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- Zensorialmente : Dejá que tu cuerpo sea tu cerebroDe EverandZensorialmente : Dejá que tu cuerpo sea tu cerebroAún no hay calificaciones
- Las Cinco Leyes Biológicas De La Naturaleza: La Nueva Medicina (Color Edition) SpanishDe EverandLas Cinco Leyes Biológicas De La Naturaleza: La Nueva Medicina (Color Edition) SpanishCalificación: 4.5 de 5 estrellas4.5/5 (3)
- Disciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreDe EverandDisciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreCalificación: 5 de 5 estrellas5/5 (3)
- Anatomía & 100 estiramientos Esenciales (Color): Técnicas, beneficios, precauciones, consejos, tablas de series, dolenciasDe EverandAnatomía & 100 estiramientos Esenciales (Color): Técnicas, beneficios, precauciones, consejos, tablas de series, dolenciasCalificación: 4.5 de 5 estrellas4.5/5 (21)
- 50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaDe Everand50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaCalificación: 4 de 5 estrellas4/5 (37)
- Batidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoDe EverandBatidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoCalificación: 5 de 5 estrellas5/5 (2)
- Interpretación del ECG: Una Guía Práctica e Intuitiva para Aprender a Leer el ECG y Diagnosticar y Tratar ArritmiasDe EverandInterpretación del ECG: Una Guía Práctica e Intuitiva para Aprender a Leer el ECG y Diagnosticar y Tratar ArritmiasAún no hay calificaciones
- Guía de cálculo y diseño de conductos para ventilación y climatizaciónDe EverandGuía de cálculo y diseño de conductos para ventilación y climatizaciónCalificación: 5 de 5 estrellas5/5 (1)
- El péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaDe EverandEl péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaCalificación: 4.5 de 5 estrellas4.5/5 (27)
- Proyectos Arduino con Tinkercad: Diseño y programación de proyectos electrónicos basados en Arduino con TinkercadDe EverandProyectos Arduino con Tinkercad: Diseño y programación de proyectos electrónicos basados en Arduino con TinkercadCalificación: 5 de 5 estrellas5/5 (1)
- La causa raiz de los accidentes: Historias de accidentes en la industriaDe EverandLa causa raiz de los accidentes: Historias de accidentes en la industriaCalificación: 5 de 5 estrellas5/5 (1)
- El concepto Mulligan de terapia manual (Color)De EverandEl concepto Mulligan de terapia manual (Color)Calificación: 5 de 5 estrellas5/5 (3)
- La vibración de las ondas cerebrales: Recuperar el ritmo de una vida saludable y felizDe EverandLa vibración de las ondas cerebrales: Recuperar el ritmo de una vida saludable y felizCalificación: 5 de 5 estrellas5/5 (7)
- Neuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaDe EverandNeuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaCalificación: 4 de 5 estrellas4/5 (16)
- Manual técnico para la interpretación de análisis de suelos y fertilización de cultivosDe EverandManual técnico para la interpretación de análisis de suelos y fertilización de cultivosCalificación: 4 de 5 estrellas4/5 (1)