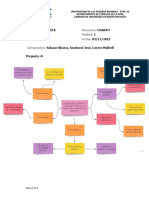
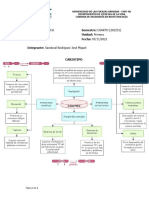
También podría gustarte
- Atlas de ciliados y otros microorganismos frecuentes en plantas de sistemas aerobio de aguas residualesDe EverandAtlas de ciliados y otros microorganismos frecuentes en plantas de sistemas aerobio de aguas residualesAún no hay calificaciones
- Práctica 2 - Analisis Filogenetico Mediante Metodo de Maxima Parsimonia y Neighbor JoiningDocumento9 páginasPráctica 2 - Analisis Filogenetico Mediante Metodo de Maxima Parsimonia y Neighbor JoiningMiguelAún no hay calificaciones
- FilogeniasDocumento4 páginasFilogeniasfelipe romeroAún no hay calificaciones
- Práctica 4Documento7 páginasPráctica 4Paula Michel Morales HernándezAún no hay calificaciones
- Realización de Un Árbol Filogenético Con El Programa MegaDocumento5 páginasRealización de Un Árbol Filogenético Con El Programa MegaMaleja Alfaro TangAún no hay calificaciones
- FG Octopoda Julian RodriguezDocumento9 páginasFG Octopoda Julian RodriguezJULIAN ALEJANDRO RODRIGUEZ URIBEAún no hay calificaciones
- Resumen Parálogos y Ortólogos.2Documento5 páginasResumen Parálogos y Ortólogos.2Lani RojasAún no hay calificaciones
- Bionformatica Taller 2Documento6 páginasBionformatica Taller 2Andres VelandiaAún no hay calificaciones
- Taller6 Constr Mi Primer Arbol FilogenetDocumento7 páginasTaller6 Constr Mi Primer Arbol FilogenetccanelounionAún no hay calificaciones
- Castro Esparza Daniel y Pérez Valenzuela Jahaziel Método BayesianoDocumento16 páginasCastro Esparza Daniel y Pérez Valenzuela Jahaziel Método BayesianoJaziel Perez ValenzuelaAún no hay calificaciones
- Informe 1 - Gonzalez - PluaDocumento9 páginasInforme 1 - Gonzalez - PluaOswaldo PluaAún no hay calificaciones
- TP16 FilogeniaDocumento2 páginasTP16 FilogeniaAlejandro IglesiasAún no hay calificaciones
- Guía 13 - Predicción y AnotaciónDocumento10 páginasGuía 13 - Predicción y AnotaciónPaula FlorezAún no hay calificaciones
- Informe Diseño de Epitopes (Inmunologia)Documento6 páginasInforme Diseño de Epitopes (Inmunologia)kevin.lopez.bAún no hay calificaciones
- Laboratorio Uso Software PopARTDocumento13 páginasLaboratorio Uso Software PopARTDania B. HerreraAún no hay calificaciones
- Cladistica 2-1Documento11 páginasCladistica 2-1MARIA CAMILA VELASCO CASTAÑEDAAún no hay calificaciones
- Analisis SecuenciasDocumento20 páginasAnalisis Secuenciasleomcm83Aún no hay calificaciones
- Filogenia MolecularDocumento33 páginasFilogenia MolecularAnghelloLozanoDíaz100% (1)
- Modelamiento Por Homología de La Enzima Aminoacil-ARNt Sintetasa Mediante El Procesador I-TasserDocumento5 páginasModelamiento Por Homología de La Enzima Aminoacil-ARNt Sintetasa Mediante El Procesador I-TasserCristianAndresGonzalezTapieroAún no hay calificaciones
- ModeloALINEAMIENTO SEQpdfDocumento15 páginasModeloALINEAMIENTO SEQpdfFrancisco FernandezAún no hay calificaciones
- Practica 1 de BiotecnologiaDocumento14 páginasPractica 1 de BiotecnologiaHector Damian FloresAún no hay calificaciones
- Biología General EncuentroFinalC12021Documento5 páginasBiología General EncuentroFinalC12021María PaulaAún no hay calificaciones
- Taller 2 PDFDocumento12 páginasTaller 2 PDFJeanpaul AcostasuarezAún no hay calificaciones
- Tema 9Documento8 páginasTema 9RobertoJuanMendezAún no hay calificaciones
- Filogenia ResumenDocumento11 páginasFilogenia ResumenMari PiliAún no hay calificaciones
- Wuolah Free EPD TEMA 3 Identificacion de Proteinas Espectometria de MasasDocumento7 páginasWuolah Free EPD TEMA 3 Identificacion de Proteinas Espectometria de MasasLuis SuarezAún no hay calificaciones
- Fase 3 - Actividad Práctica - Bases de Datos de Secuencias BiológicasDocumento16 páginasFase 3 - Actividad Práctica - Bases de Datos de Secuencias BiológicasJuan Camilo RodriguezAún no hay calificaciones
- Alineamiento y Construcción de Un Árbol Filogenético Gen Nifh (Gen de Fijación Biológica de Nitrógeno)Documento39 páginasAlineamiento y Construcción de Un Árbol Filogenético Gen Nifh (Gen de Fijación Biológica de Nitrógeno)María Fernanda Lima Totocayo100% (1)
- OtusDocumento4 páginasOtusEstefano EscanioAún no hay calificaciones
- TEMA 4 BQ eDocumento22 páginasTEMA 4 BQ eLAURA CABEZAS VINAGREAún no hay calificaciones
- ResúmenesDocumento2 páginasResúmenesFIORELLA SOFIA ZOLFI RODRIGUEZAún no hay calificaciones
- Biología General EncuentroFinalDocumento6 páginasBiología General EncuentroFinalOscar VargasAún no hay calificaciones
- FAQ en BioinformaticaDocumento12 páginasFAQ en BioinformaticaccanelounionAún no hay calificaciones
- Taller BioinformáticaDocumento10 páginasTaller Bioinformáticajose luis parraAún no hay calificaciones
- MétodoDocumento1 páginaMétodoAndres Felipe HernandezAún no hay calificaciones
- Uso de Herramientas para Alineación de Secuencias y Creación de Árboles Filogenéticos para La Determinación de EspeciesDocumento5 páginasUso de Herramientas para Alineación de Secuencias y Creación de Árboles Filogenéticos para La Determinación de EspeciesrexmonzidosAún no hay calificaciones
- AndreDocumento10 páginasAndrerodriguezlopezandreAún no hay calificaciones
- Molecular Informe 1Documento9 páginasMolecular Informe 1Gary Aldryn De La Cruz AntialonAún no hay calificaciones
- TP9 2023Documento5 páginasTP9 2023victoria ferreiraAún no hay calificaciones
- Actividad 1 - Descubriendo Un Organismo A Partir de Una Secuencia GenómicaDocumento2 páginasActividad 1 - Descubriendo Un Organismo A Partir de Una Secuencia GenómicaMarianaPocoviAún no hay calificaciones
- Taller 16 BioinformáticaDocumento6 páginasTaller 16 BioinformáticaMari PeñaAún no hay calificaciones
- Resumen ProteomicaDocumento20 páginasResumen ProteomicaAshley DudleyAún no hay calificaciones
- Como Realizar Un Árbol FilogenéticoDocumento24 páginasComo Realizar Un Árbol FilogenéticoAlejandro Cuevas Villegas100% (1)
- Informe de Bio FilogeniaDocumento20 páginasInforme de Bio FilogeniaAnyulia Torres SosaAún no hay calificaciones
- Como Realizar Un Árbol FilogenéticoDocumento24 páginasComo Realizar Un Árbol FilogenéticoAlejandro Cuevas Villegas100% (1)
- Análisis de Datos Metagenómicos (Practico-Docentes Jorge Wenzel y Andrés Iriarte) - 2017Documento4 páginasAnálisis de Datos Metagenómicos (Practico-Docentes Jorge Wenzel y Andrés Iriarte) - 2017duverney.gaviriaAún no hay calificaciones
- ENSEMBLDocumento3 páginasENSEMBLAndres GuerreroAún no hay calificaciones
- P4 - Alineamientos Múltiples - Sánchez Zamorano Julio CésarDocumento8 páginasP4 - Alineamientos Múltiples - Sánchez Zamorano Julio CésarJulio César Sánchez ZamoranoAún no hay calificaciones
- Practica N°2 Mega DnaDocumento22 páginasPractica N°2 Mega DnaHector Damian FloresAún no hay calificaciones
- Taller # 5 - Parsimonia - 2022Documento15 páginasTaller # 5 - Parsimonia - 2022MARIA CAMILA VELASCO CASTAÑEDAAún no hay calificaciones
- Lab - PCRDocumento8 páginasLab - PCRNatividad OrtegaAún no hay calificaciones
- Analisis Gen 16sDocumento11 páginasAnalisis Gen 16sValentín Ydme catacoraAún no hay calificaciones
- Alineamiento de Secuencias de Adn y Generación deDocumento10 páginasAlineamiento de Secuencias de Adn y Generación deMaria Arrazola100% (1)
- Bioinformática 29-09Documento4 páginasBioinformática 29-09バラ色いちごAún no hay calificaciones
- BC TALLER 6 Alineamientos Múltiples e Inicios en FilogeniaDocumento11 páginasBC TALLER 6 Alineamientos Múltiples e Inicios en FilogeniageraldineAún no hay calificaciones
- Teoria 3 - Bioinformatica - Luis Jaramillo PDFDocumento6 páginasTeoria 3 - Bioinformatica - Luis Jaramillo PDFMiguel Angel Alvarado RamosAún no hay calificaciones
- annotated-PRACTICA 3Documento10 páginasannotated-PRACTICA 3Andreita Estefany Ventura TejadaAún no hay calificaciones
- Copia de INFORME 1Documento5 páginasCopia de INFORME 1JuankVacaAún no hay calificaciones
- BF TALLER 6 Alineamientos Múltiples e Inicios en FilogeniaDocumento11 páginasBF TALLER 6 Alineamientos Múltiples e Inicios en FilogeniaSarah Ortiz MencoAún no hay calificaciones
- Metodología para Extracción de Semen en PsitácidosDocumento71 páginasMetodología para Extracción de Semen en PsitácidosErnesto SanchezAún no hay calificaciones
- Sandoval - Jose - Investigación 1Documento6 páginasSandoval - Jose - Investigación 1MiguelAún no hay calificaciones
- Sandoval - José - Investigación 1Documento4 páginasSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Sandoval - José - Investigación 1Documento1 páginaSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 - Unida 3Documento5 páginasSandoval - Jose - Investigación 1 - Unida 3MiguelAún no hay calificaciones
- Sandoval, Rodriguez, Investigación Bibliográfica 2. U1.Documento4 páginasSandoval, Rodriguez, Investigación Bibliográfica 2. U1.MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 1Documento1 páginaActividad en Clase Pregunta 1MiguelAún no hay calificaciones
- Variaciones Numéricas y Estructurales en Los CromosomasDocumento2 páginasVariaciones Numéricas y Estructurales en Los CromosomasMiguelAún no hay calificaciones
- Actividad en Clase Pregunta 2Documento1 páginaActividad en Clase Pregunta 2MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 3Documento1 páginaActividad en Clase Pregunta 3MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 4Documento1 páginaActividad en Clase Pregunta 4MiguelAún no hay calificaciones
- Practica 4Documento7 páginasPractica 4MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 Unidad 3Documento5 páginasSandoval - Jose - Investigación 1 Unidad 3MiguelAún no hay calificaciones
- Aplicacion Teorica de Los Postulados de Koch A Casos EspecificosDocumento3 páginasAplicacion Teorica de Los Postulados de Koch A Casos EspecificosMiguelAún no hay calificaciones
- CariotipoDocumento1 páginaCariotipoMiguelAún no hay calificaciones
- Practica 3Documento5 páginasPractica 3MiguelAún no hay calificaciones
- Práctica 4. - Procesamiento de Ficheros de TextoDocumento7 páginasPráctica 4. - Procesamiento de Ficheros de TextoMiguelAún no hay calificaciones
- Practica 1 GenesDocumento9 páginasPractica 1 GenesMiguelAún no hay calificaciones
- Pautas de Clasificacion y Taxonomia BacterianaDocumento4 páginasPautas de Clasificacion y Taxonomia BacterianaMiguelAún no hay calificaciones
- EnzimasDocumento4 páginasEnzimasMiguelAún no hay calificaciones
- Tema 18Documento26 páginasTema 18Laura AbaloAún no hay calificaciones
- El LiderazgoDocumento5 páginasEl LiderazgoNilson RamirezAún no hay calificaciones
- Tarea 2 - Elvis González - Identificación y Análisis Del Contexto LocalDocumento6 páginasTarea 2 - Elvis González - Identificación y Análisis Del Contexto LocalElvis GonzalezAún no hay calificaciones
- La Importancia de Las Pruebas Psicológicas en Las OrganizacionesDocumento3 páginasLa Importancia de Las Pruebas Psicológicas en Las Organizacionescarolina murilloAún no hay calificaciones
- Caratula Anexos 1Documento10 páginasCaratula Anexos 1Guadalupe SheylaAún no hay calificaciones
- Examen de Instrumentacion Simon Cristobal Elvis Franck 11190219Documento9 páginasExamen de Instrumentacion Simon Cristobal Elvis Franck 11190219FrankSimonCristobal100% (1)
- Marlew Af-5200 - H 2021Documento3 páginasMarlew Af-5200 - H 2021Rudy Nizama RosellAún no hay calificaciones
- Multiplicación Con RegletasDocumento7 páginasMultiplicación Con RegletasKelly Rivera riveraAún no hay calificaciones
- Articulo Tipo Informe Expociencia 2022Documento7 páginasArticulo Tipo Informe Expociencia 2022susana sierraAún no hay calificaciones
- Guía de Ejercicio1CDDocumento8 páginasGuía de Ejercicio1CDMaikito Towers Sanchez GalanAún no hay calificaciones
- QAZco 9 K 1 Ws KGTZDocumento99 páginasQAZco 9 K 1 Ws KGTZlicitacion.propuestasAún no hay calificaciones
- Trabajo de Fitopatologia 2Documento4 páginasTrabajo de Fitopatologia 2Rodrigo Bonelo GarciaAún no hay calificaciones
- Ciencia, Inventos y Experimentos en Casa Radio FMDocumento17 páginasCiencia, Inventos y Experimentos en Casa Radio FMjimmy osorioAún no hay calificaciones
- Macroprocesos CSB - UNPDocumento11 páginasMacroprocesos CSB - UNPALEXANDRA TIMANA CORDOVA100% (1)
- Proyecto Integrador Fase 2 Del Segundo Grado BDocumento18 páginasProyecto Integrador Fase 2 Del Segundo Grado BKatherine TorresAún no hay calificaciones
- Modulo 3 Actividad Integradora 1. Un Hecho HistoricoDocumento4 páginasModulo 3 Actividad Integradora 1. Un Hecho HistoricosebastianAún no hay calificaciones
- Constituye Un Sistema Integral Que Comprende Una Correlación de Partes Históricamente InmóvilesDocumento1 páginaConstituye Un Sistema Integral Que Comprende Una Correlación de Partes Históricamente Inmóvileshelio249Aún no hay calificaciones
- Peritaje o Mediacion en Los Conflictos FamiliaresDocumento10 páginasPeritaje o Mediacion en Los Conflictos FamiliaresCarla Inés Alvarado AbadAún no hay calificaciones
- Zenobio Castillo - EncuestaDocumento9 páginasZenobio Castillo - EncuestaKENJO FABRICIO ZENOBIO CASTILLOAún no hay calificaciones
- Junio - 2do Grado Educación Física (2022-2023)Documento5 páginasJunio - 2do Grado Educación Física (2022-2023)XOCHITL LUGO GILAún no hay calificaciones
- Programa CursoDocumento4 páginasPrograma CursoLuisa Fernanda GarciaAún no hay calificaciones
- Unidad 6 BioeticaDocumento39 páginasUnidad 6 BioeticaKarla Daniela Castillo SerranoAún no hay calificaciones
- WeltanschauungDocumento5 páginasWeltanschauungAlfonso Vergara OlivaAún no hay calificaciones
- U2 m04 t01 - R - TangentesDocumento5 páginasU2 m04 t01 - R - TangentesDavid Sandoval FriasAún no hay calificaciones
- Pemex RP-7Documento3 páginasPemex RP-7Said Angel Badillo Loyola C.Aún no hay calificaciones
- Ecologia 2021Documento30 páginasEcologia 2021ISABEL MMAún no hay calificaciones
- Clasificación de La AntropologíaDocumento17 páginasClasificación de La AntropologíaLeylie Vilela AlvaradoAún no hay calificaciones
- Resumen EjecutivoDocumento24 páginasResumen EjecutivoALEXANDER OSORIO MENDIETAAún no hay calificaciones
- Prob. Presion, Densidad, P. EspecificoDocumento5 páginasProb. Presion, Densidad, P. Especificopaul vsg saenz cordovaAún no hay calificaciones
- Plantilla Fase 1. Reconocimiento Del Contexto Actual Del EmprendimientoDocumento11 páginasPlantilla Fase 1. Reconocimiento Del Contexto Actual Del EmprendimientoValentina Gaitan GamezAún no hay calificaciones
- Batidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoDe EverandBatidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoCalificación: 5 de 5 estrellas5/5 (2)
- Homo antecessor: El nacimiento de una especieDe EverandHomo antecessor: El nacimiento de una especieCalificación: 5 de 5 estrellas5/5 (1)
- Inteligencia artificial: Lo que usted necesita saber sobre el aprendizaje automático, robótica, aprendizaje profundo, Internet de las cosas, redes neuronales, y nuestro futuroDe EverandInteligencia artificial: Lo que usted necesita saber sobre el aprendizaje automático, robótica, aprendizaje profundo, Internet de las cosas, redes neuronales, y nuestro futuroCalificación: 4 de 5 estrellas4/5 (1)
- Influencia. La psicología de la persuasiónDe EverandInfluencia. La psicología de la persuasiónCalificación: 4.5 de 5 estrellas4.5/5 (14)
- Cerebro y silencio: Las claves de la creatividad y la serenidadDe EverandCerebro y silencio: Las claves de la creatividad y la serenidadCalificación: 5 de 5 estrellas5/5 (2)
- Sistema de gestión lean para principiantes: Fundamentos del sistema de gestión lean para pequeñas y medianas empresas - con muchos ejemplos prácticosDe EverandSistema de gestión lean para principiantes: Fundamentos del sistema de gestión lean para pequeñas y medianas empresas - con muchos ejemplos prácticosCalificación: 4 de 5 estrellas4/5 (16)
- Cerebros rotos: Pacientes asombrosos que me enseñaron a vivirDe EverandCerebros rotos: Pacientes asombrosos que me enseñaron a vivirCalificación: 5 de 5 estrellas5/5 (3)
- 7 tendencias digitales que cambiarán el mundoDe Everand7 tendencias digitales que cambiarán el mundoCalificación: 4.5 de 5 estrellas4.5/5 (87)
- 50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaDe Everand50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaCalificación: 4 de 5 estrellas4/5 (37)
- Liberación del trauma: Perdón y temblor es el caminoDe EverandLiberación del trauma: Perdón y temblor es el caminoCalificación: 4 de 5 estrellas4/5 (4)
- El Mapa del cielo: Cómo la ciencia, la religión y la gente común están demostrando el más alláDe EverandEl Mapa del cielo: Cómo la ciencia, la religión y la gente común están demostrando el más alláCalificación: 4 de 5 estrellas4/5 (12)
- Ciencia de datos: La serie de conocimientos esenciales de MIT PressDe EverandCiencia de datos: La serie de conocimientos esenciales de MIT PressCalificación: 5 de 5 estrellas5/5 (1)
- Cultura y clima: fundamentos para el cambio en la organizaciónDe EverandCultura y clima: fundamentos para el cambio en la organizaciónAún no hay calificaciones
- Procesos de moda multifocal: Aproximaciones teóricas y prácticas sobre indumentaria latinoamericana del siglo XXIDe EverandProcesos de moda multifocal: Aproximaciones teóricas y prácticas sobre indumentaria latinoamericana del siglo XXICalificación: 3 de 5 estrellas3/5 (2)
- Historia del cuerpo humano: Evolución, salud y enfermedadDe EverandHistoria del cuerpo humano: Evolución, salud y enfermedadAún no hay calificaciones
- Sesgos Cognitivos: Una Fascinante Mirada dentro de la Psicología Humana y los Métodos para Evitar la Disonancia Cognitiva, Mejorar sus Habilidades para Resolver Problemas y Tomar Mejores DecisionesDe EverandSesgos Cognitivos: Una Fascinante Mirada dentro de la Psicología Humana y los Métodos para Evitar la Disonancia Cognitiva, Mejorar sus Habilidades para Resolver Problemas y Tomar Mejores DecisionesCalificación: 4.5 de 5 estrellas4.5/5 (13)
- El péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaDe EverandEl péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaCalificación: 4.5 de 5 estrellas4.5/5 (27)
- Excel y SQL de la mano: Trabajo con bases de datos en Excel de forma eficienteDe EverandExcel y SQL de la mano: Trabajo con bases de datos en Excel de forma eficienteCalificación: 1 de 5 estrellas1/5 (1)
- Clics contra la humanidad: Libertad y resistencia en la era de la distracción tecnológicaDe EverandClics contra la humanidad: Libertad y resistencia en la era de la distracción tecnológicaCalificación: 4.5 de 5 estrellas4.5/5 (117)
- El método Zettelkasten: Cómo tomar notas de forma eficaz para impulsar la escritura y el aprendizaje de estudiantes, académicos y escritores de no ficciónDe EverandEl método Zettelkasten: Cómo tomar notas de forma eficaz para impulsar la escritura y el aprendizaje de estudiantes, académicos y escritores de no ficciónCalificación: 4.5 de 5 estrellas4.5/5 (13)
- Guía de aplicacion de la ISO 9001:2015De EverandGuía de aplicacion de la ISO 9001:2015Calificación: 5 de 5 estrellas5/5 (3)
- 200 tareas en terapia breve: 2ª ediciónDe Everand200 tareas en terapia breve: 2ª ediciónCalificación: 4.5 de 5 estrellas4.5/5 (33)