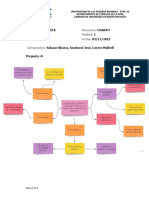
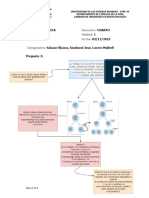
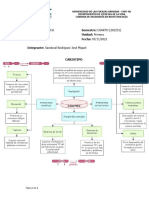
También podría gustarte
- Estocástica Teoría de ColasDocumento2 páginasEstocástica Teoría de ColasA zyzzAún no hay calificaciones
- Proyecto Del Genoma HumanoDocumento6 páginasProyecto Del Genoma HumanoAmada LopezAún no hay calificaciones
- Taller MutacionesDocumento2 páginasTaller MutacionesIsco Yepes MejiaAún no hay calificaciones
- PRACTICA 4 - Bioinformática y Curvas Estándares Con QPCR - 1Documento9 páginasPRACTICA 4 - Bioinformática y Curvas Estándares Con QPCR - 1saioa coronadoAún no hay calificaciones
- Entrevista 2Documento4 páginasEntrevista 2KEVIN PEDRO CAMARENA GAVILANAún no hay calificaciones
- MonografiaDocumento7 páginasMonografiaKiara Vanessa Bustinza Quispe100% (1)
- Practica N°2 Mega DnaDocumento22 páginasPractica N°2 Mega DnaHector Damian FloresAún no hay calificaciones
- Practica 4Documento7 páginasPractica 4MiguelAún no hay calificaciones
- Valeria Ampuero Practica N°2Documento16 páginasValeria Ampuero Practica N°2Valeria AmpueroAún no hay calificaciones
- Guion Curso APLICACIÓN DE HERRAMIENTAS BIOINFORMÁTICAS EN EL ESTUDIO DE LAS ENFERMEDADES GENÉTICAS HUMANASDocumento14 páginasGuion Curso APLICACIÓN DE HERRAMIENTAS BIOINFORMÁTICAS EN EL ESTUDIO DE LAS ENFERMEDADES GENÉTICAS HUMANASchebetomxAún no hay calificaciones
- Informe Practica n1 Emily Cusilayme RomeroDocumento16 páginasInforme Practica n1 Emily Cusilayme RomeroEmily Cusilayme RomeroAún no hay calificaciones
- Upnw 2023 - I Genoma y Salud MH1081 EapmhDocumento49 páginasUpnw 2023 - I Genoma y Salud MH1081 EapmhAnthony Baca ParejaAún no hay calificaciones
- BioestadisticaDocumento11 páginasBioestadisticaOSMERY MADALYNE VILLALVA CASTILLOAún no hay calificaciones
- Reporte BioinformáticaDocumento27 páginasReporte BioinformáticaYvan Arroyo100% (1)
- Genoma HumanoDocumento5 páginasGenoma Humanofloresmerlyn05Aún no hay calificaciones
- Biotecnología DoradaDocumento34 páginasBiotecnología DoradaRox Flores100% (1)
- Técnicas de Análisis Masivo de La Expresión de GenesDocumento4 páginasTécnicas de Análisis Masivo de La Expresión de GenesAlexander Sanchez AguilarAún no hay calificaciones
- Fundamentos Genética - MolecularDocumento5 páginasFundamentos Genética - MolecularAmilcar FermínAún no hay calificaciones
- BIOINFORMATICaDocumento23 páginasBIOINFORMATICaJuan Jose VAAún no hay calificaciones
- TAFIDocumento11 páginasTAFIFlórian JeffersonAún no hay calificaciones
- Articulo BioquimicaIIDocumento17 páginasArticulo BioquimicaIIEsteban 17Aún no hay calificaciones
- Tarea Genetica 6Documento9 páginasTarea Genetica 6mario cabezasAún no hay calificaciones
- Ciencia Omicas y Metodos de Analisis ClinicoDocumento18 páginasCiencia Omicas y Metodos de Analisis ClinicoPaulina MerchanAún no hay calificaciones
- Genesdispositivomemoria PDFDocumento6 páginasGenesdispositivomemoria PDFLuisina VazquezAún no hay calificaciones
- 01 Introducción Bases de Datos BiologicasDocumento52 páginas01 Introducción Bases de Datos BiologicasmanoloAún no hay calificaciones
- La Tecnología Del ADN en Medicina Forense - Importancia Del Indicio y Del Lugar de Los Hechos - MEDICINA FORENSE PERÚDocumento13 páginasLa Tecnología Del ADN en Medicina Forense - Importancia Del Indicio y Del Lugar de Los Hechos - MEDICINA FORENSE PERÚMariana MoralesAún no hay calificaciones
- Practica 1 de BiotecnologiaDocumento14 páginasPractica 1 de BiotecnologiaHector Damian FloresAún no hay calificaciones
- Re-10-Lab-037 Patologia II (Med) v2Documento13 páginasRe-10-Lab-037 Patologia II (Med) v2Sphinx OneAún no hay calificaciones
- Seminario Genetica Semana 3 2018 1Documento7 páginasSeminario Genetica Semana 3 2018 1Jluis Vilca ChuquipomaAún no hay calificaciones
- Informe de Diseño Primer CompletoDocumento48 páginasInforme de Diseño Primer CompletoMaleh BuitrónAún no hay calificaciones
- Biotecnologia. Mitos Sobre Los TransgenicosDocumento5 páginasBiotecnologia. Mitos Sobre Los Transgenicosoxal jayosAún no hay calificaciones
- Trabajo 3Documento8 páginasTrabajo 3Karla ViverosAún no hay calificaciones
- Clase 8 - Genómica y El Genoma HumanoDocumento135 páginasClase 8 - Genómica y El Genoma HumanoAleja DazaAún no hay calificaciones
- Practica 2 BioinformaticaDocumento4 páginasPractica 2 BioinformaticaJezCovarrubiasAún no hay calificaciones
- Informe de Biotecnologia N°3Documento20 páginasInforme de Biotecnologia N°3Hector Damian FloresAún no hay calificaciones
- Informe Biotecnologia SnapGeneDocumento13 páginasInforme Biotecnologia SnapGeneValeria AmpueroAún no hay calificaciones
- Karampetsou2014 en EsDocumento16 páginasKarampetsou2014 en EsLizbeth HernándezAún no hay calificaciones
- Lab - PCRDocumento8 páginasLab - PCRNatividad OrtegaAún no hay calificaciones
- Bases de Datos (Taller)Documento20 páginasBases de Datos (Taller)David QuinteroAún no hay calificaciones
- Ingenieria GeneticaDocumento13 páginasIngenieria GeneticaAilin GamarraAún no hay calificaciones
- Informe 7 BioinformáticaDocumento17 páginasInforme 7 BioinformáticaCatalina Merino Yunnissi0% (2)
- Taller 1 Parte A y Parte BDocumento8 páginasTaller 1 Parte A y Parte BMilagros Belen GilAún no hay calificaciones
- Guia Taller de Biologia Molecular Y Genetica Construccion Virtual de Vector de ClonamientoDocumento17 páginasGuia Taller de Biologia Molecular Y Genetica Construccion Virtual de Vector de ClonamientoNicolás Ignacio Valenzuela SayesAún no hay calificaciones
- Utilidad en La Administración de La Justicia, Repercusión Social y Aspectos ÉticosDocumento8 páginasUtilidad en La Administración de La Justicia, Repercusión Social y Aspectos ÉticosYeison Andres TobonAún no hay calificaciones
- Practica Buso y Control de Cromatogramas de Adn Utilizando El Software BioeditDocumento5 páginasPractica Buso y Control de Cromatogramas de Adn Utilizando El Software BioeditNicole Aragón ArceAún no hay calificaciones
- Genetica Forense Del Laboratorio A Los TribunalesDocumento3 páginasGenetica Forense Del Laboratorio A Los Tribunalesadrian romeroAún no hay calificaciones
- Foro 2 - IBT - Jhon YagualDocumento4 páginasForo 2 - IBT - Jhon YagualJhon Marcoss :vAún no hay calificaciones
- Proyecto Genoma HumanoDocumento17 páginasProyecto Genoma Humanoprofesormiguel241Aún no hay calificaciones
- MicroarreglosDocumento8 páginasMicroarreglospoluxivizaAún no hay calificaciones
- Terminación Del Proyecto Genoma HumanoDocumento11 páginasTerminación Del Proyecto Genoma HumanoFrankoAlexandreGamadeCoaAún no hay calificaciones
- Analisis Gen 16sDocumento11 páginasAnalisis Gen 16sValentín Ydme catacoraAún no hay calificaciones
- Genoma Humano 1Documento21 páginasGenoma Humano 1Ana Lizet Calderon AvilaAún no hay calificaciones
- Biologia de SistemasDocumento18 páginasBiologia de SistemasiredelvelAún no hay calificaciones
- Ensayo BiotecDocumento7 páginasEnsayo Biotecpsantillan2310Aún no hay calificaciones
- Informe Anual de La Persona Investigadora Contratada-Lissette Anahi Boada AcostaDocumento10 páginasInforme Anual de La Persona Investigadora Contratada-Lissette Anahi Boada AcostaLissette BoadaAún no hay calificaciones
- ADN y Medicina LegalDocumento4 páginasADN y Medicina LegalPablo ParianAún no hay calificaciones
- Quien CodificoDocumento21 páginasQuien Codificosergio Art palmerosAún no hay calificaciones
- Práctica 1 (Corregida)Documento25 páginasPráctica 1 (Corregida)JairoCris TianAún no hay calificaciones
- Guía de LaboratorioDocumento4 páginasGuía de LaboratoriocdelcidxAún no hay calificaciones
- Proyecto Genoma HumanoDocumento8 páginasProyecto Genoma HumanoLizeth Aragon GomezAún no hay calificaciones
- Sandoval - José - Investigación 1Documento1 páginaSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1Documento6 páginasSandoval - Jose - Investigación 1MiguelAún no hay calificaciones
- Sandoval, Rodriguez, Investigación Bibliográfica 2. U1.Documento4 páginasSandoval, Rodriguez, Investigación Bibliográfica 2. U1.MiguelAún no hay calificaciones
- Variaciones Numéricas y Estructurales en Los CromosomasDocumento2 páginasVariaciones Numéricas y Estructurales en Los CromosomasMiguelAún no hay calificaciones
- Sandoval - José - Investigación 1Documento4 páginasSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 - Unida 3Documento5 páginasSandoval - Jose - Investigación 1 - Unida 3MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 2Documento1 páginaActividad en Clase Pregunta 2MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 1Documento1 páginaActividad en Clase Pregunta 1MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 4Documento1 páginaActividad en Clase Pregunta 4MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 3Documento1 páginaActividad en Clase Pregunta 3MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 Unidad 3Documento5 páginasSandoval - Jose - Investigación 1 Unidad 3MiguelAún no hay calificaciones
- Practica 3Documento5 páginasPractica 3MiguelAún no hay calificaciones
- CariotipoDocumento1 páginaCariotipoMiguelAún no hay calificaciones
- Prueba Analisis FilogeneticoDocumento9 páginasPrueba Analisis FilogeneticoMiguelAún no hay calificaciones
- Practica 1 GenesDocumento9 páginasPractica 1 GenesMiguelAún no hay calificaciones
- Aplicacion Teorica de Los Postulados de Koch A Casos EspecificosDocumento3 páginasAplicacion Teorica de Los Postulados de Koch A Casos EspecificosMiguelAún no hay calificaciones
- Pautas de Clasificacion y Taxonomia BacterianaDocumento4 páginasPautas de Clasificacion y Taxonomia BacterianaMiguelAún no hay calificaciones
- EnzimasDocumento4 páginasEnzimasMiguelAún no hay calificaciones
- Ciencias 2Documento4 páginasCiencias 2LordDreamAún no hay calificaciones
- Dolor de EstomagoDocumento3 páginasDolor de EstomagoBryan Gallardo MartínezAún no hay calificaciones
- Accion de Tutela. AccionDocumento15 páginasAccion de Tutela. AccionSteven TorresAún no hay calificaciones
- Reinos Guia 1Documento2 páginasReinos Guia 1Marcela BuitragoAún no hay calificaciones
- Hay Más de Una Forma de Cultivar TilapiaDocumento8 páginasHay Más de Una Forma de Cultivar TilapiaAngeloAún no hay calificaciones
- Tecnica 5W+2HDocumento3 páginasTecnica 5W+2HLuis Arturo Zuta KomoriAún no hay calificaciones
- TELETRABAJODocumento2 páginasTELETRABAJOorlando mamaniAún no hay calificaciones
- Fármacos Hipouricemiantes y AntigotososDocumento29 páginasFármacos Hipouricemiantes y AntigotososJair ZamcazAún no hay calificaciones
- Trabajo Mecánico en Caliente y en FríoDocumento86 páginasTrabajo Mecánico en Caliente y en FríoPollo LozanoAún no hay calificaciones
- Adonde Fueron Los Bichos PDFDocumento23 páginasAdonde Fueron Los Bichos PDFElizabeth MarlboroughAún no hay calificaciones
- Trazo de Vestido de Licra para DamaDocumento8 páginasTrazo de Vestido de Licra para DamaKaren Liliana Pérez CastillejoAún no hay calificaciones
- Military Review Los NIveles de La Guerra Como Niveles de Analisis (Nov 22)Documento7 páginasMilitary Review Los NIveles de La Guerra Como Niveles de Analisis (Nov 22)Raul Ivan Ramos PeraltaAún no hay calificaciones
- Diagramas de Árbol y Regla de BayesDocumento4 páginasDiagramas de Árbol y Regla de BayesrosaAún no hay calificaciones
- Deber Practica Experimental 5 de GestiónDocumento6 páginasDeber Practica Experimental 5 de GestiónJorge VargasAún no hay calificaciones
- Almacenamiento de MercanciasDocumento7 páginasAlmacenamiento de MercanciasJherson Leiva RafaelAún no hay calificaciones
- Integrales HiperbólicasDocumento13 páginasIntegrales HiperbólicasÓsmarAcurio100% (1)
- Cuadro ComparativoDocumento2 páginasCuadro ComparativoSebastianAún no hay calificaciones
- 021agrupar Desagrupar AutoesquemasDocumento91 páginas021agrupar Desagrupar AutoesquemasjssjjsjsAún no hay calificaciones
- Libro Fundamentos de Ing de Yacimientos - Fredy EscobarDocumento570 páginasLibro Fundamentos de Ing de Yacimientos - Fredy EscobarJulieta Vila ZambranaAún no hay calificaciones
- Puno 21 6Documento22 páginasPuno 21 6carlos_mamani_19Aún no hay calificaciones
- Keto RecetasDocumento11 páginasKeto RecetasPablo CastilloAún no hay calificaciones
- PUD #3 9º Noveno MatematicaDocumento10 páginasPUD #3 9º Noveno MatematicaPaulinoAún no hay calificaciones
- Hechos de Armas Bajo La Bandera de Álvaro MutisDocumento2 páginasHechos de Armas Bajo La Bandera de Álvaro MutisDiego Valverde VillenaAún no hay calificaciones
- Fisioterapia Aplicada A Pacientes Geriátricos Con Patología TraumatoDocumento5 páginasFisioterapia Aplicada A Pacientes Geriátricos Con Patología TraumatoCëecy DiazAún no hay calificaciones
- NMX C 159 Onncce 2016 Curado y Met de MoldearDocumento17 páginasNMX C 159 Onncce 2016 Curado y Met de MoldearEdgar Joel Perez Carvajal100% (1)
- Gravimetria HierroDocumento6 páginasGravimetria HierroLuis Eduardo RenteriaAún no hay calificaciones
- MontaplatosDocumento16 páginasMontaplatosgiAún no hay calificaciones
- Manual de Producción de CamoteDocumento25 páginasManual de Producción de CamoteMiguel CáceresAún no hay calificaciones