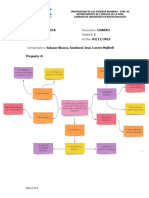
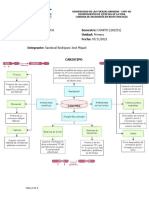
También podría gustarte
- Atlas de ciliados y otros microorganismos frecuentes en plantas de sistemas aerobio de aguas residualesDe EverandAtlas de ciliados y otros microorganismos frecuentes en plantas de sistemas aerobio de aguas residualesAún no hay calificaciones
- Prueba Analisis FilogeneticoDocumento9 páginasPrueba Analisis FilogeneticoMiguelAún no hay calificaciones
- Realización de Un Árbol Filogenético Con El Programa MegaDocumento5 páginasRealización de Un Árbol Filogenético Con El Programa MegaMaleja Alfaro TangAún no hay calificaciones
- Práctica 4Documento7 páginasPráctica 4Paula Michel Morales HernándezAún no hay calificaciones
- FilogeniasDocumento4 páginasFilogeniasfelipe romeroAún no hay calificaciones
- FG Octopoda Julian RodriguezDocumento9 páginasFG Octopoda Julian RodriguezJULIAN ALEJANDRO RODRIGUEZ URIBEAún no hay calificaciones
- Resumen Parálogos y Ortólogos.2Documento5 páginasResumen Parálogos y Ortólogos.2Lani RojasAún no hay calificaciones
- Bionformatica Taller 2Documento6 páginasBionformatica Taller 2Andres VelandiaAún no hay calificaciones
- Castro Esparza Daniel y Pérez Valenzuela Jahaziel Método BayesianoDocumento16 páginasCastro Esparza Daniel y Pérez Valenzuela Jahaziel Método BayesianoJaziel Perez ValenzuelaAún no hay calificaciones
- Taller6 Constr Mi Primer Arbol FilogenetDocumento7 páginasTaller6 Constr Mi Primer Arbol FilogenetccanelounionAún no hay calificaciones
- Guía 13 - Predicción y AnotaciónDocumento10 páginasGuía 13 - Predicción y AnotaciónPaula FlorezAún no hay calificaciones
- Informe 1 - Gonzalez - PluaDocumento9 páginasInforme 1 - Gonzalez - PluaOswaldo PluaAún no hay calificaciones
- Informe Diseño de Epitopes (Inmunologia)Documento6 páginasInforme Diseño de Epitopes (Inmunologia)kevin.lopez.bAún no hay calificaciones
- TP16 FilogeniaDocumento2 páginasTP16 FilogeniaAlejandro IglesiasAún no hay calificaciones
- ModeloALINEAMIENTO SEQpdfDocumento15 páginasModeloALINEAMIENTO SEQpdfFrancisco FernandezAún no hay calificaciones
- Taller 2 PDFDocumento12 páginasTaller 2 PDFJeanpaul AcostasuarezAún no hay calificaciones
- Cladistica 2-1Documento11 páginasCladistica 2-1MARIA CAMILA VELASCO CASTAÑEDAAún no hay calificaciones
- Filogenia MolecularDocumento33 páginasFilogenia MolecularAnghelloLozanoDíaz100% (1)
- Biología General EncuentroFinalC12021Documento5 páginasBiología General EncuentroFinalC12021María PaulaAún no hay calificaciones
- OtusDocumento4 páginasOtusEstefano EscanioAún no hay calificaciones
- Laboratorio Uso Software PopARTDocumento13 páginasLaboratorio Uso Software PopARTDania B. HerreraAún no hay calificaciones
- Analisis SecuenciasDocumento20 páginasAnalisis Secuenciasleomcm83Aún no hay calificaciones
- Filogenia ResumenDocumento11 páginasFilogenia ResumenMari PiliAún no hay calificaciones
- Tema 9Documento8 páginasTema 9RobertoJuanMendezAún no hay calificaciones
- ResúmenesDocumento2 páginasResúmenesFIORELLA SOFIA ZOLFI RODRIGUEZAún no hay calificaciones
- Biología General EncuentroFinalDocumento6 páginasBiología General EncuentroFinalOscar VargasAún no hay calificaciones
- Fase 3 - Actividad Práctica - Bases de Datos de Secuencias BiológicasDocumento16 páginasFase 3 - Actividad Práctica - Bases de Datos de Secuencias BiológicasJuan Camilo RodriguezAún no hay calificaciones
- Wuolah Free EPD TEMA 3 Identificacion de Proteinas Espectometria de MasasDocumento7 páginasWuolah Free EPD TEMA 3 Identificacion de Proteinas Espectometria de MasasLuis SuarezAún no hay calificaciones
- AndreDocumento10 páginasAndrerodriguezlopezandreAún no hay calificaciones
- Modelamiento Por Homología de La Enzima Aminoacil-ARNt Sintetasa Mediante El Procesador I-TasserDocumento5 páginasModelamiento Por Homología de La Enzima Aminoacil-ARNt Sintetasa Mediante El Procesador I-TasserCristianAndresGonzalezTapieroAún no hay calificaciones
- Taller BioinformáticaDocumento10 páginasTaller Bioinformáticajose luis parraAún no hay calificaciones
- Actividad 1 - Descubriendo Un Organismo A Partir de Una Secuencia GenómicaDocumento2 páginasActividad 1 - Descubriendo Un Organismo A Partir de Una Secuencia GenómicaMarianaPocoviAún no hay calificaciones
- Practica 1 de BiotecnologiaDocumento14 páginasPractica 1 de BiotecnologiaHector Damian FloresAún no hay calificaciones
- Alineamiento y Construcción de Un Árbol Filogenético Gen Nifh (Gen de Fijación Biológica de Nitrógeno)Documento39 páginasAlineamiento y Construcción de Un Árbol Filogenético Gen Nifh (Gen de Fijación Biológica de Nitrógeno)María Fernanda Lima Totocayo100% (1)
- TEMA 4 BQ eDocumento22 páginasTEMA 4 BQ eLAURA CABEZAS VINAGREAún no hay calificaciones
- Molecular Informe 1Documento9 páginasMolecular Informe 1Gary Aldryn De La Cruz AntialonAún no hay calificaciones
- TP9 2023Documento5 páginasTP9 2023victoria ferreiraAún no hay calificaciones
- Como Realizar Un Árbol FilogenéticoDocumento24 páginasComo Realizar Un Árbol FilogenéticoAlejandro Cuevas Villegas100% (1)
- MétodoDocumento1 páginaMétodoAndres Felipe HernandezAún no hay calificaciones
- FAQ en BioinformaticaDocumento12 páginasFAQ en BioinformaticaccanelounionAún no hay calificaciones
- Taller 16 BioinformáticaDocumento6 páginasTaller 16 BioinformáticaMari PeñaAún no hay calificaciones
- Como Realizar Un Árbol FilogenéticoDocumento24 páginasComo Realizar Un Árbol FilogenéticoAlejandro Cuevas Villegas100% (1)
- Lab - PCRDocumento8 páginasLab - PCRNatividad OrtegaAún no hay calificaciones
- Bioinformática 29-09Documento4 páginasBioinformática 29-09バラ色いちごAún no hay calificaciones
- Análisis de Datos Metagenómicos (Practico-Docentes Jorge Wenzel y Andrés Iriarte) - 2017Documento4 páginasAnálisis de Datos Metagenómicos (Practico-Docentes Jorge Wenzel y Andrés Iriarte) - 2017duverney.gaviriaAún no hay calificaciones
- Uso de Herramientas para Alineación de Secuencias y Creación de Árboles Filogenéticos para La Determinación de EspeciesDocumento5 páginasUso de Herramientas para Alineación de Secuencias y Creación de Árboles Filogenéticos para La Determinación de EspeciesrexmonzidosAún no hay calificaciones
- Resumen ProteomicaDocumento20 páginasResumen ProteomicaAshley DudleyAún no hay calificaciones
- ENSEMBLDocumento3 páginasENSEMBLAndres GuerreroAún no hay calificaciones
- annotated-PRACTICA 3Documento10 páginasannotated-PRACTICA 3Andreita Estefany Ventura TejadaAún no hay calificaciones
- MetagenomicaDocumento50 páginasMetagenomicaErik LugoAún no hay calificaciones
- Metodología para Extracción de Semen en PsitácidosDocumento71 páginasMetodología para Extracción de Semen en PsitácidosErnesto SanchezAún no hay calificaciones
- Alineamiento Multiple Tipo ClustalDocumento27 páginasAlineamiento Multiple Tipo ClustalValeria Gutierrez RuizAún no hay calificaciones
- Bases Datos 2013Documento43 páginasBases Datos 2013faustohernanAún no hay calificaciones
- Trabajo EMBOSTDocumento7 páginasTrabajo EMBOSTsebman89Aún no hay calificaciones
- Ejercicios Paso 3 Mejoramiento Animal 2020Documento9 páginasEjercicios Paso 3 Mejoramiento Animal 2020Yeni BareñoAún no hay calificaciones
- Caicedo & Sarria, Proyecto Final BromelioideaeDocumento5 páginasCaicedo & Sarria, Proyecto Final BromelioideaeAlba Lucía Caicedo MuñozAún no hay calificaciones
- Ciencias OmicasDocumento3 páginasCiencias OmicasARIZBETH GUADALUPE VILLANUEVA FLORESAún no hay calificaciones
- BC TALLER 6 Alineamientos Múltiples e Inicios en FilogeniaDocumento11 páginasBC TALLER 6 Alineamientos Múltiples e Inicios en FilogeniageraldineAún no hay calificaciones
- Informe de Biotecnologia N°3Documento20 páginasInforme de Biotecnologia N°3Hector Damian FloresAún no hay calificaciones
- Taller # 5 - Parsimonia - 2022Documento15 páginasTaller # 5 - Parsimonia - 2022MARIA CAMILA VELASCO CASTAÑEDAAún no hay calificaciones
- Sandoval - Jose - Investigación 1Documento6 páginasSandoval - Jose - Investigación 1MiguelAún no hay calificaciones
- Sandoval, Rodriguez, Investigación Bibliográfica 2. U1.Documento4 páginasSandoval, Rodriguez, Investigación Bibliográfica 2. U1.MiguelAún no hay calificaciones
- Sandoval - José - Investigación 1Documento1 páginaSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Variaciones Numéricas y Estructurales en Los CromosomasDocumento2 páginasVariaciones Numéricas y Estructurales en Los CromosomasMiguelAún no hay calificaciones
- Sandoval - José - Investigación 1Documento4 páginasSandoval - José - Investigación 1MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 4Documento1 páginaActividad en Clase Pregunta 4MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 - Unida 3Documento5 páginasSandoval - Jose - Investigación 1 - Unida 3MiguelAún no hay calificaciones
- Sandoval - Jose - Investigación 1 Unidad 3Documento5 páginasSandoval - Jose - Investigación 1 Unidad 3MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 3Documento1 páginaActividad en Clase Pregunta 3MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 1Documento1 páginaActividad en Clase Pregunta 1MiguelAún no hay calificaciones
- Actividad en Clase Pregunta 2Documento1 páginaActividad en Clase Pregunta 2MiguelAún no hay calificaciones
- Practica 4Documento7 páginasPractica 4MiguelAún no hay calificaciones
- Aplicacion Teorica de Los Postulados de Koch A Casos EspecificosDocumento3 páginasAplicacion Teorica de Los Postulados de Koch A Casos EspecificosMiguelAún no hay calificaciones
- Practica 1 GenesDocumento9 páginasPractica 1 GenesMiguelAún no hay calificaciones
- Practica 3Documento5 páginasPractica 3MiguelAún no hay calificaciones
- Práctica 4. - Procesamiento de Ficheros de TextoDocumento7 páginasPráctica 4. - Procesamiento de Ficheros de TextoMiguelAún no hay calificaciones
- CariotipoDocumento1 páginaCariotipoMiguelAún no hay calificaciones
- Pautas de Clasificacion y Taxonomia BacterianaDocumento4 páginasPautas de Clasificacion y Taxonomia BacterianaMiguelAún no hay calificaciones
- EnzimasDocumento4 páginasEnzimasMiguelAún no hay calificaciones
- Calendario Ambiental 2021Documento4 páginasCalendario Ambiental 2021Larry Oruro GonzalesAún no hay calificaciones
- CALENDARIOSDocumento2 páginasCALENDARIOSCristina Del PradoAún no hay calificaciones
- Test Es Usted NegociadorDocumento5 páginasTest Es Usted NegociadorStephany VillanuevaAún no hay calificaciones
- Comparación de Flashes en OdontologíaDocumento2 páginasComparación de Flashes en OdontologíaVictor LappostAún no hay calificaciones
- Tema 1-Introducción Al Estudio de La PresupuestaciónDocumento14 páginasTema 1-Introducción Al Estudio de La PresupuestaciónAbdiel Tavárez100% (1)
- Utn - TestingDocumento3 páginasUtn - TestingBetoAún no hay calificaciones
- Carlos HellerDocumento21 páginasCarlos HellerLuis Gustavo MartinezAún no hay calificaciones
- Sem.01 Analizamos La Participacion Democratica en La Sociedad Antigua Primer AñoDocumento4 páginasSem.01 Analizamos La Participacion Democratica en La Sociedad Antigua Primer AñoCalebGYAún no hay calificaciones
- El AdverbioDocumento13 páginasEl AdverbioEsdrasRiosAún no hay calificaciones
- Unidad 6 ElectromagnetismoDocumento9 páginasUnidad 6 ElectromagnetismojoseAún no hay calificaciones
- Guía de Las Plantas Medicinales - Información de 200 Plantas Medicinales, Sus Propiedades e Indicaciones (Volumen #1) (Spanish Edition)Documento410 páginasGuía de Las Plantas Medicinales - Información de 200 Plantas Medicinales, Sus Propiedades e Indicaciones (Volumen #1) (Spanish Edition)Angie Lomeli100% (10)
- 08 - Ortografía de Las TildesDocumento6 páginas08 - Ortografía de Las TildesJholberth AvalosAún no hay calificaciones
- Pensamiento Lógico - EcuRedDocumento3 páginasPensamiento Lógico - EcuRedSophie ElizabethAún no hay calificaciones
- Rol AsistencialDocumento3 páginasRol AsistencialJavier RamirezAún no hay calificaciones
- Puntos de PellizcoDocumento1 páginaPuntos de PellizcooliverbpeAún no hay calificaciones
- Supervision EducativaDocumento6 páginasSupervision EducativaMaii delgadoAún no hay calificaciones
- Calculo Mecánico en Vanos Desnivelados PDFDocumento16 páginasCalculo Mecánico en Vanos Desnivelados PDFZtǾnee Arnie TenǾrio MǾntesAún no hay calificaciones
- Trabajogrupal, Presupuesto de Ventas.Documento11 páginasTrabajogrupal, Presupuesto de Ventas.Nataly SosaAún no hay calificaciones
- Introducción A La Ingeniería de Minera Vol. IV LAs Funciones de La Ingeniería MineraDocumento46 páginasIntroducción A La Ingeniería de Minera Vol. IV LAs Funciones de La Ingeniería MineraDIOMEDES YUNIOR CHAMORRO MONAGOAún no hay calificaciones
- Brochure de Promoción y Prevención 2020-0723Documento2 páginasBrochure de Promoción y Prevención 2020-0723Jenifer RamirezAún no hay calificaciones
- Problemas de Optimizacion y Razon de CambioDocumento3 páginasProblemas de Optimizacion y Razon de CambioHelena Dulcey H.Aún no hay calificaciones
- Examen de Ciencias Naturales 2° SemestreDocumento7 páginasExamen de Ciencias Naturales 2° SemestreVeronica GuerreroAún no hay calificaciones
- 7 2 Teoria ColasDocumento30 páginas7 2 Teoria ColasRigobertoAlexandValladaresAún no hay calificaciones
- Homomorfismo de Grupos PDFDocumento14 páginasHomomorfismo de Grupos PDFvqt2000100% (1)
- EdMe Strohmaier - Al BiruniDocumento8 páginasEdMe Strohmaier - Al BiruniAngel Chavez EslavaAún no hay calificaciones
- Arme y Desarme de Un Motor ElectricoDocumento6 páginasArme y Desarme de Un Motor ElectricosemagorAún no hay calificaciones
- Los Informes de ProcesoDocumento2 páginasLos Informes de Procesoherman Van de Velde0% (1)
- Acfrogb Crldtiaeff8spvpeiema4 CsDocumento9 páginasAcfrogb Crldtiaeff8spvpeiema4 CsJairo Antonio Becerra CastroAún no hay calificaciones
- Diagrama de Operaciones - Brazo HidráulicoDocumento1 páginaDiagrama de Operaciones - Brazo HidráulicoVictorAún no hay calificaciones
- Ciencias 8 Basico, 16 Al 20 de AgostoDocumento1 páginaCiencias 8 Basico, 16 Al 20 de AgostoSandra Morales GuerraAún no hay calificaciones