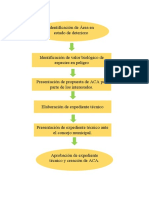
También podría gustarte
- Encuesta Residuos SolidosDocumento4 páginasEncuesta Residuos SolidosNancy Espinoza BravoAún no hay calificaciones
- Marco Teórico de Agentes QuímicosDocumento3 páginasMarco Teórico de Agentes QuímicosAndrea Llanas100% (1)
- 001 - Reyes Silva David Gonzalo - 74469087Documento24 páginas001 - Reyes Silva David Gonzalo - 74469087David Gonzalo Reyes SilvaAún no hay calificaciones
- Informe Biotecnologia SnapGeneDocumento13 páginasInforme Biotecnologia SnapGeneValeria AmpueroAún no hay calificaciones
- Genética Del Color OvinosDocumento3 páginasGenética Del Color OvinosJair Silva Dela RosaAún no hay calificaciones
- Laboratorio N 8 Estabilidad Oxidativa Por El Metodo RancimatDocumento17 páginasLaboratorio N 8 Estabilidad Oxidativa Por El Metodo RancimatRonaldAún no hay calificaciones
- Muestreo de Aguas ResidualesDocumento8 páginasMuestreo de Aguas ResidualesLya MéndezAún no hay calificaciones
- Reglamento de Grados y Titulos v12!07!2021 Final-1626817542Documento40 páginasReglamento de Grados y Titulos v12!07!2021 Final-1626817542susanaAún no hay calificaciones
- 05 - Calculo NMP-TablaDocumento4 páginas05 - Calculo NMP-TablaRoAún no hay calificaciones
- Sanitizante A Base de Cuaternario de Amonio Precaución: NO. CAS: 68424-95-3Documento43 páginasSanitizante A Base de Cuaternario de Amonio Precaución: NO. CAS: 68424-95-3Noe RojasAún no hay calificaciones
- Lista Chequeo Factores de RiesgoDocumento5 páginasLista Chequeo Factores de RiesgoAnonymous G9mkL3lhPAún no hay calificaciones
- Cuadro Sinóptico IbnorcaDocumento1 páginaCuadro Sinóptico IbnorcaSebastian Zárate VilelaAún no hay calificaciones
- Importancia de Microorganismos en La AgroindustriaDocumento4 páginasImportancia de Microorganismos en La AgroindustriaAlejandra Rengifo Tamayo0% (1)
- Plan de Contingencia - Espacio Confinado - JediomDocumento9 páginasPlan de Contingencia - Espacio Confinado - JediomBRAYAN ANTHONY MEDRANO COCHACHIAún no hay calificaciones
- Tesis Obtención de Licor de Sauco Por DestilaciónDocumento173 páginasTesis Obtención de Licor de Sauco Por Destilaciónedwin ramos zavalaAún no hay calificaciones
- Guia Taller Bioindicadores Del Agua - Copia-1Documento2 páginasGuia Taller Bioindicadores Del Agua - Copia-1Sandra CrosstrainingAún no hay calificaciones
- Cuestionario ResueltoDocumento9 páginasCuestionario ResueltoSebastian ValleAún no hay calificaciones
- Microsoft Word - Tabla AlimentosDocumento1 páginaMicrosoft Word - Tabla AlimentosOliviaAyaneguiAún no hay calificaciones
- Informe Medios de CultivoDocumento2 páginasInforme Medios de CultivoJHON ALBERT GODOY GOMEZAún no hay calificaciones
- Sistema Bioclimático HoldridgeDocumento11 páginasSistema Bioclimático HoldridgeKaren ChasiAún no hay calificaciones
- BiologiaDocumento5 páginasBiologiaROBERT ANDRES FUENTES NIÑOAún no hay calificaciones
- 2,3,5Documento12 páginas2,3,5Fabricio Nuñez100% (1)
- Informe de Laboratorio Lacteos Grupo 2.2 JuevesDocumento34 páginasInforme de Laboratorio Lacteos Grupo 2.2 JuevesAndrés BonillaAún no hay calificaciones
- Practica n4 LecheDocumento6 páginasPractica n4 LecheYandel AlvarezAún no hay calificaciones
- Agentes y Causas de La DeforestacionDocumento14 páginasAgentes y Causas de La DeforestacionLucía MartinezAún no hay calificaciones
- La Pirámide de Control de Riesgos de BirdDocumento5 páginasLa Pirámide de Control de Riesgos de BirdAnderson Villarreal SiguenzaAún no hay calificaciones
- Aspergillus FumigatusDocumento1 páginaAspergillus FumigatusAnderson AlvaradoAún no hay calificaciones
- Control y Prevencion de La Contaminacion Industrial en Fabricacion de Productos LacteosDocumento57 páginasControl y Prevencion de La Contaminacion Industrial en Fabricacion de Productos LacteosLuis Alfonso Hidalgo TamayoAún no hay calificaciones
- Despacho Viceministerial de Desarrollo e Infraestructura Agraria y RiegoDocumento28 páginasDespacho Viceministerial de Desarrollo e Infraestructura Agraria y RiegoCristy GarcíaAún no hay calificaciones
- FT Cloro GranuladoDocumento1 páginaFT Cloro GranuladomarybelAún no hay calificaciones
- Ceras en AlimentosDocumento2 páginasCeras en AlimentosIsrael ArboleydaAún no hay calificaciones
- Poe-001-Induccion A La BioseguridadDocumento3 páginasPoe-001-Induccion A La BioseguridadLeoncio CornelioAún no hay calificaciones
- Visita Al Sistema Hídrico Del Embalse de Pasto Grande...Documento23 páginasVisita Al Sistema Hídrico Del Embalse de Pasto Grande...Diego fernandoAún no hay calificaciones
- Grasa Litio Ep-2Documento3 páginasGrasa Litio Ep-2luis armandoAún no hay calificaciones
- Dicromato de PotasioDocumento14 páginasDicromato de PotasioCynthia Zarate VasquezAún no hay calificaciones
- Refriger Ac I OnDocumento103 páginasRefriger Ac I OnPitufina DianitaAún no hay calificaciones
- Resultados. Microscopia y MicrometríaDocumento4 páginasResultados. Microscopia y MicrometríaMelisa RubioAún no hay calificaciones
- Determinacion de Puntos Criticos.Documento2 páginasDeterminacion de Puntos Criticos.Scarleth PalaciosAún no hay calificaciones
- Informe Gramineas y LeguminosasDocumento13 páginasInforme Gramineas y LeguminosasFernanda SinchiAún no hay calificaciones
- Pigars 2015 - 2025Documento184 páginasPigars 2015 - 2025Anccas Wil AnccasAún no hay calificaciones
- Fisiologia VegetalDocumento11 páginasFisiologia Vegetalcarlostk16Aún no hay calificaciones
- Diagnostico Cuenca Endorreica BoliviaDocumento11 páginasDiagnostico Cuenca Endorreica BoliviaClau BordaAún no hay calificaciones
- Sinopsis Cronológica de La Biotecnología Ambiental Desde Sus OrígenesDocumento7 páginasSinopsis Cronológica de La Biotecnología Ambiental Desde Sus OrígenesJoshy Alarcon M.Aún no hay calificaciones
- Diagrama de IshikawaDocumento1 páginaDiagrama de IshikawaAngela AbarcaAún no hay calificaciones
- Aprovechamiento de Subproductos de La Faena de Aves. Curtido de Pieles de Pata de GallinaDocumento7 páginasAprovechamiento de Subproductos de La Faena de Aves. Curtido de Pieles de Pata de GallinaDiana Sofía CaipeAún no hay calificaciones
- Actividad I Microbiologia AmbientalDocumento8 páginasActividad I Microbiologia Ambientaljjsuescun24100% (1)
- BBME U1 EA MAAS - PlataformaDocumento10 páginasBBME U1 EA MAAS - PlataformaMartha Maria Araiza SantiniAún no hay calificaciones
- Rio CitarumDocumento1 páginaRio CitarumJHOEL LLANOS ALVARADOAún no hay calificaciones
- Protocolo de Seguridad para El Laboratorio de Tecnologia en Frutas y HortalizasDocumento17 páginasProtocolo de Seguridad para El Laboratorio de Tecnologia en Frutas y HortalizasJuanGarciaBenavidesAún no hay calificaciones
- Matriz de Autoevaluación - 34 EstandaresDocumento9 páginasMatriz de Autoevaluación - 34 EstandaresJakc PhvaAún no hay calificaciones
- Proceso de Faenado de PollosDocumento5 páginasProceso de Faenado de Pollosjuanvladimir87Aún no hay calificaciones
- MONOGRAFÍADocumento23 páginasMONOGRAFÍAMartin Carrasco AlarconAún no hay calificaciones
- Separacion y Purificación de GasesDocumento4 páginasSeparacion y Purificación de GasesjairoAún no hay calificaciones
- Aplicacion de Los Carbohidratos en El AmbienteDocumento3 páginasAplicacion de Los Carbohidratos en El AmbientePatricio OrozcoAún no hay calificaciones
- Cosecha y Comercializacion de La QuinuaDocumento24 páginasCosecha y Comercializacion de La QuinuaJorge LuisAún no hay calificaciones
- Informe 1 Normas de Seguridad en El Laboratorio de BiologíaDocumento4 páginasInforme 1 Normas de Seguridad en El Laboratorio de BiologíaGary ChoqueAún no hay calificaciones
- Valeria Ampuero Practica N°2Documento16 páginasValeria Ampuero Practica N°2Valeria AmpueroAún no hay calificaciones
- Practica 1 de BiotecnologiaDocumento14 páginasPractica 1 de BiotecnologiaHector Damian FloresAún no hay calificaciones
- Analisis Gen 16sDocumento11 páginasAnalisis Gen 16sValentín Ydme catacoraAún no hay calificaciones
- Informe de Biotecnologia N°3Documento20 páginasInforme de Biotecnologia N°3Hector Damian FloresAún no hay calificaciones
- Ley de Fourier, DaltonDocumento14 páginasLey de Fourier, DaltonHector Damian FloresAún no hay calificaciones
- Tarea 3 Terminada de EstadisticaDocumento1 páginaTarea 3 Terminada de EstadisticaHector Damian FloresAún no hay calificaciones
- Biologia de Suelos Fonte PDFDocumento36 páginasBiologia de Suelos Fonte PDFCamilo Torre CamposAún no hay calificaciones
- Reactores IndustrialesDocumento19 páginasReactores IndustrialesHector Damian FloresAún no hay calificaciones
- Reactores IndustrialesDocumento19 páginasReactores IndustrialesHector Damian FloresAún no hay calificaciones
- Tarea 3 Terminada de EstadisticaDocumento1 páginaTarea 3 Terminada de EstadisticaHector Damian FloresAún no hay calificaciones
- METODO DEL FlOTADOR AVANCEDocumento9 páginasMETODO DEL FlOTADOR AVANCEHector Damian FloresAún no hay calificaciones
- Tablero en BlancoDocumento1 páginaTablero en BlancoHector Damian FloresAún no hay calificaciones
- Análisis de Textura....Documento4 páginasAnálisis de Textura....Hector Damian FloresAún no hay calificaciones
- Grupo EnvironmentDocumento5 páginasGrupo EnvironmentHector Damian FloresAún no hay calificaciones
- Rio HuarmeyDocumento22 páginasRio HuarmeyHector Damian FloresAún no hay calificaciones
- Rio HuarmeyDocumento50 páginasRio HuarmeyHector Damian FloresAún no hay calificaciones
- Rio HuarmeyDocumento22 páginasRio HuarmeyHector Damian FloresAún no hay calificaciones
- PROPUESTA DE ACA Lomas de Amoquinto-GESTION AMBIENTALDocumento30 páginasPROPUESTA DE ACA Lomas de Amoquinto-GESTION AMBIENTALHector Damian FloresAún no hay calificaciones
- GanaderiaDocumento49 páginasGanaderiaHector Damian FloresAún no hay calificaciones
- Flu Jog RamaDocumento1 páginaFlu Jog RamaHector Damian FloresAún no hay calificaciones
- GRUPO 1 Recursos NaturalesDocumento4 páginasGRUPO 1 Recursos NaturalesHector Damian FloresAún no hay calificaciones
- Ejercicio Flujo de CajaDocumento3 páginasEjercicio Flujo de CajaHector Damian FloresAún no hay calificaciones
- Iso 14000 - Grupo - 1Documento21 páginasIso 14000 - Grupo - 1Hector Damian FloresAún no hay calificaciones
- Estructura Del AdnDocumento17 páginasEstructura Del AdnHector Damian FloresAún no hay calificaciones
- Mpacto Social y Económico en El Uso de Biocombustibles DiapositivasDocumento1 páginaMpacto Social y Económico en El Uso de Biocombustibles DiapositivasHector Damian FloresAún no hay calificaciones
- 2Documento5 páginas2Hector Damian FloresAún no hay calificaciones
- Resumen Aguas1.1Documento1 páginaResumen Aguas1.1Hector Damian FloresAún no hay calificaciones
- Marco Teórico: Contaminación Del Agua MarinaDocumento11 páginasMarco Teórico: Contaminación Del Agua MarinaHector Damian FloresAún no hay calificaciones
- RESUMEN... Aguas .....Documento1 páginaRESUMEN... Aguas .....Hector Damian FloresAún no hay calificaciones
- Tablero en BlancoDocumento1 páginaTablero en BlancoHector Damian FloresAún no hay calificaciones
- Informe de Biotecnologia N°3Documento20 páginasInforme de Biotecnologia N°3Hector Damian FloresAún no hay calificaciones
- Universidad Nacional de MoqueguaDocumento2 páginasUniversidad Nacional de MoqueguaHector Damian FloresAún no hay calificaciones
- El Área de Conservación AmbientalDocumento1 páginaEl Área de Conservación AmbientalHector Damian FloresAún no hay calificaciones
- Link Del InformeDocumento1 páginaLink Del InformeHector Damian FloresAún no hay calificaciones
- ConversaciónDocumento23 páginasConversaciónwakinepadreAún no hay calificaciones
- Antropología MexicanaDocumento24 páginasAntropología MexicanaJessica DíazAún no hay calificaciones
- 2METODOLOGÍA - VILLANUEVA RODRÍGUEZ - 2dosemestre - ELAJOLOTEDocumento9 páginas2METODOLOGÍA - VILLANUEVA RODRÍGUEZ - 2dosemestre - ELAJOLOTEMaría Fernanda Villanueva RodríguezAún no hay calificaciones
- Gobierno CorporativoDocumento10 páginasGobierno Corporativocelki mariela hernandez quispeAún no hay calificaciones
- Patricia Lindo y Fabiola Amariles. 2015. Metodologías de Evaluación: Voces de Cambio en La Vida de Las MujeresDocumento18 páginasPatricia Lindo y Fabiola Amariles. 2015. Metodologías de Evaluación: Voces de Cambio en La Vida de Las MujeresFabiola Amariles-AutoraAún no hay calificaciones
- Geología AplicadaDocumento9 páginasGeología AplicadaGabriela Jonaily Urdaneta QuijadaAún no hay calificaciones
- Plan Anual 2022Documento9 páginasPlan Anual 2022Hernan AstradaAún no hay calificaciones
- Test Psicometricos.Documento6 páginasTest Psicometricos.Alejandro HernándezAún no hay calificaciones
- Silabo de PlaneamientoDocumento9 páginasSilabo de PlaneamientoFranciscoli Alex Ppacco Huamani0% (1)
- Tarea 3Documento3 páginasTarea 3Jerome YapulAún no hay calificaciones
- Unidad 3 Tarea 4 Calidaddevida ActividadindividualDocumento7 páginasUnidad 3 Tarea 4 Calidaddevida ActividadindividualWilson Daza PachecoAún no hay calificaciones
- Ejercicios L3Documento2 páginasEjercicios L3Rockruto 33Aún no hay calificaciones
- Biorregiones de La TierraDocumento6 páginasBiorregiones de La Tierraalfredoatiencia50% (2)
- Antividad 11 Unidad 4 Adm General Del Procesador Ejercicio PlanifiDocumento1 páginaAntividad 11 Unidad 4 Adm General Del Procesador Ejercicio PlanifiAntonioAún no hay calificaciones
- ODONTOPEDIATRIADocumento136 páginasODONTOPEDIATRIAandreaAún no hay calificaciones
- Meme UAIDocumento30 páginasMeme UAIantonio rebolledoAún no hay calificaciones
- Li2208 Quick Start Guide EsDocumento2 páginasLi2208 Quick Start Guide EscamilaAún no hay calificaciones
- Tutorial PRA NEAEDocumento9 páginasTutorial PRA NEAEib1326Aún no hay calificaciones
- TareaS05.Gutierrez Putpaña - Diego EstefanoDocumento1 páginaTareaS05.Gutierrez Putpaña - Diego EstefanoDiego EstefanoAún no hay calificaciones
- Ark4 - Ea1 - Inicial 2 2021Documento20 páginasArk4 - Ea1 - Inicial 2 2021Gigi TrujilloAún no hay calificaciones
- Manual CEMUCAF PDFDocumento12 páginasManual CEMUCAF PDFJuan Pablo CastroAún no hay calificaciones
- Examen de Suficiencia Por Culminacion de Carrera - Riveros PoloDocumento10 páginasExamen de Suficiencia Por Culminacion de Carrera - Riveros PoloangelAún no hay calificaciones
- Plan de Trabajo 2023Documento15 páginasPlan de Trabajo 2023Mileny Chinchay Inga100% (1)
- Fase de ControlDocumento12 páginasFase de ControlDaniel Ramos GómezAún no hay calificaciones
- Módulo 05 - Sesión 04 - GerontologíaDocumento45 páginasMódulo 05 - Sesión 04 - Gerontologíacynthia garcia mamaniAún no hay calificaciones
- Ejercicios MinimizacionDocumento4 páginasEjercicios MinimizacionJose Manuel Molina CuariteAún no hay calificaciones
- Control de AccesoDocumento30 páginasControl de Accesocarlos0% (1)
- Topografia 1 y Laboratorio Tarea 9Documento7 páginasTopografia 1 y Laboratorio Tarea 9Felix Jose Altagracia EspinalAún no hay calificaciones
- Pertenecer Al Universo Fritjof Capra PDF 11 3Documento3 páginasPertenecer Al Universo Fritjof Capra PDF 11 3Jesus Pérez cAún no hay calificaciones
- Planeacion Semana 18 Sexto GradoDocumento14 páginasPlaneacion Semana 18 Sexto GradoYeseniaAún no hay calificaciones