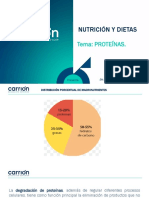
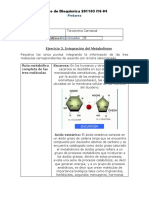
También podría gustarte
- Ayudas ergogénicas y nutricionalesDe EverandAyudas ergogénicas y nutricionalesCalificación: 5 de 5 estrellas5/5 (2)
- Tema13 Catabolismo AminoacidosDocumento36 páginasTema13 Catabolismo AminoacidosÁngela Díaz ChamorroAún no hay calificaciones
- Identificación de azúcares reductores con reactivo de FehlingDocumento9 páginasIdentificación de azúcares reductores con reactivo de FehlingManuel PeñaAún no hay calificaciones
- Metabolismo de AminoacidosDocumento3 páginasMetabolismo de AminoacidosErikaDesimoniAún no hay calificaciones
- Síntesis y Degradación de AminoácidosDocumento4 páginasSíntesis y Degradación de AminoácidosLorena ArroyoAún no hay calificaciones
- Metabolismo de Los AminoácidosDocumento4 páginasMetabolismo de Los AminoácidosGuillermo Jose PerezAún no hay calificaciones
- Metabolismo de AminoácidosDocumento12 páginasMetabolismo de AminoácidosGermán Sil100% (1)
- Metabolismo de AminoacidosDocumento3 páginasMetabolismo de AminoacidosAntonella BeratzAún no hay calificaciones
- Metabolismo de Las ProteínasDocumento40 páginasMetabolismo de Las ProteínasFrank Sánchez Estela100% (1)
- Degradación de Compuestos Nitrogenados.Documento19 páginasDegradación de Compuestos Nitrogenados.Zulema Velázquez GarcíaAún no hay calificaciones
- Sem 12 File 3Documento26 páginasSem 12 File 3Daniela MatamalaAún no hay calificaciones
- Trabalho de Bioquimica 2Documento11 páginasTrabalho de Bioquimica 2Thayssa De Souza Silva 3GAún no hay calificaciones
- AminotransferasaDocumento10 páginasAminotransferasaAmgel FloresAún no hay calificaciones
- Bio Qui MicaDocumento9 páginasBio Qui MicaJatnielAún no hay calificaciones
- Metabolismo Proteínas-EstDocumento32 páginasMetabolismo Proteínas-Estgerson1contreras-1Aún no hay calificaciones
- AAs: Transaminación, Desaminación y Pool de AAsDocumento14 páginasAAs: Transaminación, Desaminación y Pool de AAsAlexander SanchezAún no hay calificaciones
- HJHJJJKDocumento15 páginasHJHJJJKSanti GarciaAún no hay calificaciones
- Metabolismo de AminoacidosDocumento13 páginasMetabolismo de AminoacidosDiego Fidelli LopezAún no hay calificaciones
- Catabolismo de Los AminoácidosDocumento39 páginasCatabolismo de Los AminoácidosEd BozaAún no hay calificaciones
- Resumen Proteinas y AminoacidosDocumento30 páginasResumen Proteinas y Aminoacidosmarianne beltranAún no hay calificaciones
- Desaminacion TransaminacionDocumento10 páginasDesaminacion TransaminacionHector Manuel Lendo HernandezAún no hay calificaciones
- El Pool de AminoácidosDocumento9 páginasEl Pool de AminoácidosKarla Torres100% (1)
- Metabolismo ProteinasDocumento9 páginasMetabolismo ProteinasTania CabreraAún no hay calificaciones
- Funciones Clave de La Peptidil SerinaDocumento8 páginasFunciones Clave de La Peptidil SerinaAG PCAún no hay calificaciones
- RESUMEN. Metabolismo de Los Compuestos NitrogenadosDocumento8 páginasRESUMEN. Metabolismo de Los Compuestos NitrogenadosFernanda AraizaAún no hay calificaciones
- 3 Metabolismo de Proteinas 2016Documento11 páginas3 Metabolismo de Proteinas 2016Estela EscalanteAún no hay calificaciones
- Clase 11 Metabolismo ProteinasDocumento46 páginasClase 11 Metabolismo ProteinasyaipeneAún no hay calificaciones
- NUTRICION Y DIETAS. Semana 9Documento17 páginasNUTRICION Y DIETAS. Semana 9cristina delia chombo cutipaAún no hay calificaciones
- Clases Unidad 6 201820Documento51 páginasClases Unidad 6 201820DiegoAún no hay calificaciones
- TextoDocumento4 páginasTextoAriana SilvaAún no hay calificaciones
- Seminario 10 Catabolismo de AminoácidosDocumento5 páginasSeminario 10 Catabolismo de AminoácidosVictor CespedesAún no hay calificaciones
- Reporte Sobre MetabolismoDocumento3 páginasReporte Sobre MetabolismoKatherine YisselAún no hay calificaciones
- Metabolismo de AA - Resumen Qca IIDocumento10 páginasMetabolismo de AA - Resumen Qca IIVEROAún no hay calificaciones
- LA ALIMENTACION Y NUTRICION EN POLIGASTRICOS Examen 2Documento17 páginasLA ALIMENTACION Y NUTRICION EN POLIGASTRICOS Examen 2ManuelAún no hay calificaciones
- Tema 11 Bioquímica, FBDocumento40 páginasTema 11 Bioquímica, FBRODRIGO CORTESAún no hay calificaciones
- Corrección General Tarea 8 Metabolismo de NitrogenadosDocumento14 páginasCorrección General Tarea 8 Metabolismo de NitrogenadosAnto MandarinoAún no hay calificaciones
- Proteolisis AminoacidosDocumento5 páginasProteolisis AminoacidosLuis SantesAún no hay calificaciones
- AA GlucogénicosDocumento2 páginasAA GlucogénicosGabriela PrimorAún no hay calificaciones
- Metabolismo de ProteínasDocumento57 páginasMetabolismo de ProteínasAndrea CondoAún no hay calificaciones
- Metabolismo Glucólisis (ALUMNOS 3)Documento22 páginasMetabolismo Glucólisis (ALUMNOS 3)danielaestefaniasalinasrojas8Aún no hay calificaciones
- Compuestos NitrogenadosDocumento26 páginasCompuestos NitrogenadosKaren Veronica Garzon SalazarAún no hay calificaciones
- NitrogenoDocumento8 páginasNitrogenoLili VanAún no hay calificaciones
- Digestión y Absorción de Las ProteínasDocumento2 páginasDigestión y Absorción de Las Proteínaskaty yesenia apaza charccahuanaAún no hay calificaciones
- Teórico 13 Dra. Rosenstein - Metabolismo de Aminoácidos - 2018Documento24 páginasTeórico 13 Dra. Rosenstein - Metabolismo de Aminoácidos - 2018Nandy PachecoAún no hay calificaciones
- 5. Metabolismo de Proteinas ResumenDocumento77 páginas5. Metabolismo de Proteinas ResumenIris zavalaAún no hay calificaciones
- Transaminación y Desaminación de ProteínasDocumento24 páginasTransaminación y Desaminación de ProteínasCITLALLI MU�OZ ANDRADEAún no hay calificaciones
- Resumen Unidad 6 201820Documento4 páginasResumen Unidad 6 201820Fernanda BecerraAún no hay calificaciones
- Introduccion Al MetabolismoDocumento11 páginasIntroduccion Al MetabolismoMelissa CazaresAún no hay calificaciones
- Metabolismo Del Nitrogeno. DegradacionDocumento29 páginasMetabolismo Del Nitrogeno. DegradacionFernando Rodriguez ChavezAún no hay calificaciones
- Introducción Al Metabolismo de Los AminoácidosDocumento27 páginasIntroducción Al Metabolismo de Los AminoácidosmadeleynecyAún no hay calificaciones
- Resuemen de BQM Metabolismo y Degradación de AADocumento2 páginasResuemen de BQM Metabolismo y Degradación de AAOldemar GarciaAún no hay calificaciones
- T13 Reacciones PDFDocumento17 páginasT13 Reacciones PDFcarlos esteban diaz aragonAún no hay calificaciones
- Bioquimica 2Documento7 páginasBioquimica 2shaq20011100% (3)
- Ejercicio2 - Tarea 3 - Grupo ColaborativoDocumento7 páginasEjercicio2 - Tarea 3 - Grupo ColaborativoJuliana SalazarAún no hay calificaciones
- Metabolismo de Las ProteínasDocumento14 páginasMetabolismo de Las ProteínasMaria Andrea CorredorAún no hay calificaciones
- Metabolismo de Compuestos NitrogenadosDocumento5 páginasMetabolismo de Compuestos NitrogenadosJonathan Atlahua MorenoAún no hay calificaciones
- Caminos Metabolicos ComunesDocumento30 páginasCaminos Metabolicos ComunesFelipe Monsalve IdrovoAún no hay calificaciones
- Metabolismo de Aminoácidos: Prof: Nerio SocorroDocumento47 páginasMetabolismo de Aminoácidos: Prof: Nerio SocorroMildriana RiveroAún no hay calificaciones
- Ciclo de KrebsDocumento8 páginasCiclo de KrebsMilka Rafaela Acero Vargas50% (2)
- Protein AsDocumento29 páginasProtein AsGabii PachecoAún no hay calificaciones
- Sardina tcm30-102530Documento2 páginasSardina tcm30-102530Cristhian LópezAún no hay calificaciones
- Síntesis de AG y ColesterolDocumento37 páginasSíntesis de AG y ColesterolTrisAún no hay calificaciones
- Proteinas 4 MedioDocumento39 páginasProteinas 4 MedioSumikoAún no hay calificaciones
- Fisiopatología Coagulacion PDFDocumento67 páginasFisiopatología Coagulacion PDFCarlos Mario SotoAún no hay calificaciones
- Informe EnzimasDocumento17 páginasInforme EnzimasJunior Andrade JaqueAún no hay calificaciones
- Taller Bioquimica #1-2020Documento3 páginasTaller Bioquimica #1-2020Alexander Echeverri RuizAún no hay calificaciones
- Apuntes Metabolismo de LípidosDocumento13 páginasApuntes Metabolismo de LípidosDanae Scarlet CeferinoAún no hay calificaciones
- Acidos Grasos EsencialesDocumento36 páginasAcidos Grasos EsencialesNicolMendozaAún no hay calificaciones
- Enzimas 3Documento8 páginasEnzimas 3Aly NpAún no hay calificaciones
- Metabolismo vitaminasDocumento4 páginasMetabolismo vitaminasJohana AlpalaAún no hay calificaciones
- Bioquimica en Proceso KLDocumento21 páginasBioquimica en Proceso KLAlejandra RojasAún no hay calificaciones
- Hoja Trabajo Capítulo 17Documento9 páginasHoja Trabajo Capítulo 17Sarita Fong18Aún no hay calificaciones
- Síntesis y Ciclo de La UreaDocumento2 páginasSíntesis y Ciclo de La UreaLuz Del Alba Tirado LizAún no hay calificaciones
- EICOSANOIDESDocumento17 páginasEICOSANOIDESFatima QuintanaAún no hay calificaciones
- Metabolismo de Proteinas 1Documento54 páginasMetabolismo de Proteinas 1v cvdsxdefAún no hay calificaciones
- Clase Teórica #1 - Biomoléculas-2C-2023 TN-ViviDocumento80 páginasClase Teórica #1 - Biomoléculas-2C-2023 TN-ViviRenata CuttittaAún no hay calificaciones
- EJERCICIO 3. Comprimido PDFDocumento2 páginasEJERCICIO 3. Comprimido PDFKilgor HerreraAún no hay calificaciones
- La Desnaturalización de La ProteinaDocumento2 páginasLa Desnaturalización de La ProteinaValen aranaAún no hay calificaciones
- Carbohidratos: clasificación, funciones y orígenesDocumento2 páginasCarbohidratos: clasificación, funciones y orígenesMiguel Valencia TorresAún no hay calificaciones
- Enmanuel Bioquimica IIDocumento25 páginasEnmanuel Bioquimica IIDiego Guevara ParraAún no hay calificaciones
- NewLab4 BiocompuestosDocumento8 páginasNewLab4 BiocompuestosSebastián MontesAún no hay calificaciones
- Quimica Bioinorganica de Los Elementos No-MetalicosDocumento40 páginasQuimica Bioinorganica de Los Elementos No-MetalicosFREILEN BRAYAN HUANCA CUSIAún no hay calificaciones
- Transcripcion GeneticaDocumento19 páginasTranscripcion GeneticaCristhianVilelaTinedoAún no hay calificaciones
- Bioquimica Estructural y MetabólicaDocumento158 páginasBioquimica Estructural y MetabólicaBryanLuzón100% (1)
- Macromoléculas: Caracterización por Ensayos ColorimétricosDocumento8 páginasMacromoléculas: Caracterización por Ensayos ColorimétricosDavid Solar OliveraAún no hay calificaciones
- Ejercicio Guía 1 Ejercicio 8Documento2 páginasEjercicio Guía 1 Ejercicio 8Gabi De FilippisAún no hay calificaciones
- Informe de PCRDocumento9 páginasInforme de PCRLeo Llatas Llúncor67% (3)
- Composición Química de La SABILADocumento2 páginasComposición Química de La SABILAFabricio Almendras63% (8)
- Enzimas y coenzimas: catalizadores biológicosDocumento28 páginasEnzimas y coenzimas: catalizadores biológicosCitlali PeñaAún no hay calificaciones