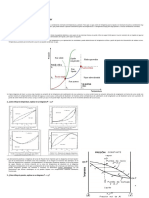
También podría gustarte
- Casos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSDe EverandCasos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSCalificación: 4.5 de 5 estrellas4.5/5 (7)
- TREMODINAMICADocumento23 páginasTREMODINAMICAmanuel100% (3)
- Raoult RecopilacionDocumento22 páginasRaoult RecopilacionMaría Argomedo Reyes100% (1)
- Ley de Raoult ModificadaDocumento8 páginasLey de Raoult ModificadaMilagrosCabanillasInfantesAún no hay calificaciones
- Equilibrio Liquido-VaporDocumento10 páginasEquilibrio Liquido-VaporMiguelAún no hay calificaciones
- Modelamiento y casos especiales de la cinética química heterogéneaDe EverandModelamiento y casos especiales de la cinética química heterogéneaCalificación: 3 de 5 estrellas3/5 (1)
- Ejercicios Semana 7 Fisioquimica PDFDocumento7 páginasEjercicios Semana 7 Fisioquimica PDFRociio GutiierrezAún no hay calificaciones
- Psicometría. Principios básicos y protocolos experimentales diversosDe EverandPsicometría. Principios básicos y protocolos experimentales diversosAún no hay calificaciones
- Volatilidad RelativaDocumento6 páginasVolatilidad RelativaAna Milena Castañeda Villamarin100% (1)
- Ley de Raoult y HenryDocumento4 páginasLey de Raoult y HenrySofia Cabarcas100% (1)
- ENTROPIADocumento17 páginasENTROPIAJonathan Pablo AntonioAún no hay calificaciones
- Guia de Estudios 1Documento7 páginasGuia de Estudios 1Alfredo AlfaroAún no hay calificaciones
- Practica 4 Lab Int 2Documento14 páginasPractica 4 Lab Int 2YulissaAún no hay calificaciones
- Informe de Laboratorio #04Documento20 páginasInforme de Laboratorio #04ruttAún no hay calificaciones
- Op1 Pre1Documento5 páginasOp1 Pre1FernandoAún no hay calificaciones
- Equilibrio Líquido Vapor en Un Sistema No IdealDocumento3 páginasEquilibrio Líquido Vapor en Un Sistema No IdealDeyaniraAún no hay calificaciones
- Practica 1 AspenDocumento22 páginasPractica 1 AspenFercho Flores SuárezAún no hay calificaciones
- 1 Preexamen Termodinamica de Procesos IIDocumento24 páginas1 Preexamen Termodinamica de Procesos IIPaul Ruiz BaldeonAún no hay calificaciones
- Modelos de ActividadDocumento56 páginasModelos de Actividadluis cordobaAún no hay calificaciones
- La FugocidadDocumento9 páginasLa FugocidadaddddddddfffffAún no hay calificaciones
- Equilibrio LV PDFDocumento14 páginasEquilibrio LV PDFJefanny JaouhariAún no hay calificaciones
- Práctica de AzeotropoDocumento10 páginasPráctica de AzeotropoJuan Sosa San GermanAún no hay calificaciones
- Resumen Procesos de Separación Capitulo 4Documento3 páginasResumen Procesos de Separación Capitulo 4Morelia MartínezAún no hay calificaciones
- p4. Lab Termo 1Documento15 páginasp4. Lab Termo 1Castillo Pérez PaulinaAún no hay calificaciones
- Ingeneria Quimica ComputacionalDocumento18 páginasIngeneria Quimica ComputacionalMirellea AndurayAún no hay calificaciones
- HOLLAND C. FundamentosModelado-EvaporadoresDocumento23 páginasHOLLAND C. FundamentosModelado-EvaporadoresJared CMAún no hay calificaciones
- LOpU1 Pre1 GascónCastilloAsafJoshuaDocumento4 páginasLOpU1 Pre1 GascónCastilloAsafJoshuaAsaf Joshua Gascón CastilloAún no hay calificaciones
- TERMODocumento23 páginasTERMOlorenaAún no hay calificaciones
- Ley de Raoult - Ecuacion de AntoineDocumento7 páginasLey de Raoult - Ecuacion de AntoineDoris JacintoAún no hay calificaciones
- Equilibrio de Fase Líquido - VaporDocumento28 páginasEquilibrio de Fase Líquido - VaporIvan TorresAún no hay calificaciones
- Equilibrio de FasesDocumento5 páginasEquilibrio de FasesLeOnel FherNandoAún no hay calificaciones
- Balances de Materia y EntalpíaDocumento5 páginasBalances de Materia y EntalpíaJavier AlbaAún no hay calificaciones
- PsdsimpDocumento6 páginasPsdsimpJósmar Jesús PérezAún no hay calificaciones
- Coeficiente de Actividad de LiquidosDocumento31 páginasCoeficiente de Actividad de LiquidosAnonymous aXbHjNyAún no hay calificaciones
- Pre1 OperacionesDocumento6 páginasPre1 OperacionesIngeniera Pau JerezAún no hay calificaciones
- Resumen - Procesos de Separación Vapor LíquidoDocumento11 páginasResumen - Procesos de Separación Vapor LíquidoNICOLAS CASTELLANOS TAMAYOAún no hay calificaciones
- Informe de Termodinámica Liquido VaporDocumento13 páginasInforme de Termodinámica Liquido VaporStiven K. Rosales CasasAún no hay calificaciones
- Unidad I DestilaciónDocumento32 páginasUnidad I DestilaciónKAREN ESTEFANíA HERNáNDEZ OCAMPOAún no hay calificaciones
- Coeficiente de Actividad de LiquidosDocumento31 páginasCoeficiente de Actividad de LiquidosConsueloRodriguez50% (2)
- Lecciones de RepasoDocumento18 páginasLecciones de RepasoRicardoAún no hay calificaciones
- Elv Etanol AguaDocumento18 páginasElv Etanol AguaJuan Montalvo CerronAún no hay calificaciones
- Cuestionario Soluciones Liq LiqDocumento1 páginaCuestionario Soluciones Liq LiqAna Gabriela González MartínezAún no hay calificaciones
- El Modelo de Dos Líquidos No AleatoriaDocumento2 páginasEl Modelo de Dos Líquidos No Aleatoriayuresama0% (1)
- Equilibrio Liquido-Vapor Metanol-TiofenoDocumento8 páginasEquilibrio Liquido-Vapor Metanol-TiofenoAaLee IsaisAún no hay calificaciones
- Tarea 3.3 equilibrioOODocumento8 páginasTarea 3.3 equilibrioOOAlex Mendoza AyaviriAún no hay calificaciones
- Ecuaciones de EstadoDocumento4 páginasEcuaciones de Estadoda1031soAún no hay calificaciones
- Propiedades de Las Sustancias PurasDocumento7 páginasPropiedades de Las Sustancias PurasYordan Andres Saez PetroAún no hay calificaciones
- Informe Práctica #3 Punto de BurbujaDocumento5 páginasInforme Práctica #3 Punto de BurbujaDiego Nicolas ManceraAún no hay calificaciones
- Tarea2 Jhoyosb JsmorenolaDocumento11 páginasTarea2 Jhoyosb JsmorenolaJuan David Hoyos BohorquezAún no hay calificaciones
- Practica 6 Lab SSFDocumento12 páginasPractica 6 Lab SSFAndie Flores0% (1)
- PosterDocumento1 páginaPosterGiseth Bibiana Sastoque AgudeloAún no hay calificaciones
- Equilibrio Líquido Vapor en Un Sistema No IdealDocumento5 páginasEquilibrio Líquido Vapor en Un Sistema No IdealAnonymous ykiLcGa4wAún no hay calificaciones
- Practica 1 Lab Opiii (Equilibrio Líquido-Vapor y Diagramas de Equilibrio)Documento18 páginasPractica 1 Lab Opiii (Equilibrio Líquido-Vapor y Diagramas de Equilibrio)vidaurre_89Aún no hay calificaciones
- Modelo GammaDocumento22 páginasModelo GammaReticular ConsciousnessAún no hay calificaciones
- 5.5 El Cambio de Entropía de Sustancias PurasDocumento4 páginas5.5 El Cambio de Entropía de Sustancias PurasrobertoAún no hay calificaciones
- Practica 4Documento6 páginasPractica 4Giulianna RodriguezAún no hay calificaciones
- PROYECTODocumento10 páginasPROYECTOLalommoreno Moreno OvandoAún no hay calificaciones
- Transferencia de Masa Ii - Tarea - N°1 - Grupo5Documento13 páginasTransferencia de Masa Ii - Tarea - N°1 - Grupo5HEVONNY JACKELINE TEJADA CALLEAún no hay calificaciones
- Copia de Práctica 3 y 4 - EH 2015Documento26 páginasCopia de Práctica 3 y 4 - EH 2015angelriosvazquez08Aún no hay calificaciones
- 03 DestilacionDocumento14 páginas03 DestilacionLuis MolloAún no hay calificaciones
- ABSORCIONDocumento29 páginasABSORCIONRut SuárezAún no hay calificaciones
- Diferencia Entre Gas y VaporDocumento8 páginasDiferencia Entre Gas y VaporJosé TorrezAún no hay calificaciones
- Practica 4 Termo de FasesDocumento32 páginasPractica 4 Termo de FasesYeraldin AviñaAún no hay calificaciones
- Ensayo Residuos SolidosDocumento3 páginasEnsayo Residuos SolidosAlitzel SarabiaAún no hay calificaciones
- UntitledDocumento1 páginaUntitledAlitzel SarabiaAún no hay calificaciones
- Transferencia de MasaDocumento9 páginasTransferencia de MasaAlitzel SarabiaAún no hay calificaciones
- Clasificación Del Residuo Peso de Las Bolsas (KG) Peso de Los RSU (KG) Porcentaje Del Subproducto Peso Volumétrico VolumenDocumento1 páginaClasificación Del Residuo Peso de Las Bolsas (KG) Peso de Los RSU (KG) Porcentaje Del Subproducto Peso Volumétrico VolumenAlitzel SarabiaAún no hay calificaciones
- GUÍA DE RESPUESTA EN CASO DE EMERGENCIA Dicloruro EtilfosfonotioicoDocumento12 páginasGUÍA DE RESPUESTA EN CASO DE EMERGENCIA Dicloruro EtilfosfonotioicoAlitzel SarabiaAún no hay calificaciones
- Normas Mas Utilizadas en La Industria QuimicaDocumento9 páginasNormas Mas Utilizadas en La Industria QuimicaAlitzel SarabiaAún no hay calificaciones
- Atlas de Riesgo-ProducciónDocumento2 páginasAtlas de Riesgo-ProducciónAlitzel SarabiaAún no hay calificaciones
- Funciones de Los PlanosDocumento2 páginasFunciones de Los PlanosAlitzel Sarabia100% (2)
- Unidad 4. Q.analíticaDocumento55 páginasUnidad 4. Q.analíticaAlitzel SarabiaAún no hay calificaciones
- Introduccion Destilacion SimpleDocumento1 páginaIntroduccion Destilacion SimpleAlitzel SarabiaAún no hay calificaciones
- Destilación Simple - AzeótroposDocumento2 páginasDestilación Simple - AzeótroposSuny StylesAún no hay calificaciones
- Modelos de SolucionDocumento5 páginasModelos de SolucionDaniel Gonzales ZayasAún no hay calificaciones
- Capitulo X - Corregido Equilibrio de FasesDocumento19 páginasCapitulo X - Corregido Equilibrio de FasesJose davidAún no hay calificaciones
- Informe #1 - Equilibrio Liquido-VaporDocumento17 páginasInforme #1 - Equilibrio Liquido-VaporJeanette Huaya RamosAún no hay calificaciones
- TESINADocumento43 páginasTESINACarmen MedinaAún no hay calificaciones
- Informe 9 Equilibrio Liquido VaporDocumento24 páginasInforme 9 Equilibrio Liquido VaporPIERO MATIAS HERRERA SUAREZAún no hay calificaciones
- Silabo - Físico QuímicaDocumento6 páginasSilabo - Físico QuímicaCristopher SaldañaAún no hay calificaciones
- Practica #2 Lineas de EquilibrioDocumento15 páginasPractica #2 Lineas de EquilibrioErikaCorderoLópezAún no hay calificaciones
- Equilibrio Líquido-Vapor en Sistemas Liquidos BinariosDocumento7 páginasEquilibrio Líquido-Vapor en Sistemas Liquidos BinarioslupitaAún no hay calificaciones
- Destilacion FinalDocumento15 páginasDestilacion FinalValeria Delgado Chávez50% (2)
- Ley RaoultDocumento9 páginasLey RaoultOriel CastroAún no hay calificaciones
- La Ley de RaoultDocumento11 páginasLa Ley de RaoultViviana FarfanAún no hay calificaciones
- Webconferencia 4 761Documento26 páginasWebconferencia 4 761Fabian CordobaAún no hay calificaciones
- Conferencia 01 Termodinámica QuímicaDocumento12 páginasConferencia 01 Termodinámica QuímicaLisselysAún no hay calificaciones
- Tema 05Documento50 páginasTema 05Fernando ModregoAún no hay calificaciones
- 4.-Ecuaciones de Estado - Cálculo de Propiedades Con Ecuaciones de EstadoDocumento34 páginas4.-Ecuaciones de Estado - Cálculo de Propiedades Con Ecuaciones de Estadokagf8750% (2)
- Ley de RaoultDocumento6 páginasLey de RaoultYessi LopzAún no hay calificaciones
- Webconferencia 8 764Documento30 páginasWebconferencia 8 764Camilo NiñoAún no hay calificaciones
- Ejemplo - Informe 9Documento16 páginasEjemplo - Informe 9Xflk lsbtAún no hay calificaciones
- Teoria de La Evaporacion - InvestigaciónDocumento10 páginasTeoria de La Evaporacion - InvestigaciónManuelAún no hay calificaciones
- Unidad I Fisicoquimica II 2020Documento23 páginasUnidad I Fisicoquimica II 2020ariel yana morgaAún no hay calificaciones
- Informe de Laboratorio Procesos 4Documento21 páginasInforme de Laboratorio Procesos 4Guillermo BaqueroAún no hay calificaciones
- Ley de RoultDocumento20 páginasLey de RoultAlexis HuancaAún no hay calificaciones
- Soluciones Propiedades ColigativasDocumento75 páginasSoluciones Propiedades ColigativasXiomara ACAún no hay calificaciones
- Ley de RauoltDocumento13 páginasLey de RauoltPaolo Guarniz TufinioAún no hay calificaciones
- Practica 3Documento7 páginasPractica 3Garcia Barrera EuniceAún no hay calificaciones