También podría gustarte
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleAún no hay calificaciones
- Sindrome de MarfanDocumento9 páginasSindrome de MarfanJohn Milton López GonzálezAún no hay calificaciones
- Sindrome de MarfánDocumento26 páginasSindrome de MarfánIsabel SevillaAún no hay calificaciones
- Presentacion Sindrome de MarfanDocumento16 páginasPresentacion Sindrome de MarfanCAJA NACIONAL DE SALUD BIOLOGIA MOLECULARAún no hay calificaciones
- Síndrome de MarfanDocumento8 páginasSíndrome de MarfanJesus NinaAún no hay calificaciones
- Sindrome de MarfanDocumento20 páginasSindrome de MarfanOscar IvanAún no hay calificaciones
- Síndrome de MarfanDocumento3 páginasSíndrome de MarfanAlicia GonzálezAún no hay calificaciones
- Síndrome de Marfan - Publicaciones CientíficasDocumento9 páginasSíndrome de Marfan - Publicaciones CientíficasmonicagalassoAún no hay calificaciones
- Síndrome de MarfanDocumento14 páginasSíndrome de MarfanRosa PúrpuraAún no hay calificaciones
- Síndrome de Marfan: enfermedad rara del tejido conectivoDocumento4 páginasSíndrome de Marfan: enfermedad rara del tejido conectivoDaniel ColmenarezAún no hay calificaciones
- Síndrome de MarfanDocumento3 páginasSíndrome de MarfanRachel HuberAún no hay calificaciones
- PatologíasDocumento25 páginasPatologíasALMA RIVERA HERNANDEZAún no hay calificaciones
- Síndrome de MarfanDocumento14 páginasSíndrome de MarfanzurypalafAún no hay calificaciones
- Síndromes más frecuentes en EcuadorDocumento6 páginasSíndromes más frecuentes en EcuadorChristian BermeoAún no hay calificaciones
- GUIA GENETICA ABERR. CROM AUTOSDocumento16 páginasGUIA GENETICA ABERR. CROM AUTOScarolinaweide21Aún no hay calificaciones
- Autosómica Dominante.2007.PostgradoDocumento58 páginasAutosómica Dominante.2007.PostgradoDiamelys MuchachoAún no hay calificaciones
- Sindrome de Marfan DietaDocumento26 páginasSindrome de Marfan Dietajose miguel aguilar saucedoAún no hay calificaciones
- Trabajo de MalformacionesDocumento18 páginasTrabajo de Malformacionesmelanie.tunonAún no hay calificaciones
- MarfanDocumento9 páginasMarfanGolvin MartínezAún no hay calificaciones
- 12 Enf Ad M IIIDocumento79 páginas12 Enf Ad M IIIYinna GonzalezAún no hay calificaciones
- Síndrome de Marfan SEMINARIODocumento63 páginasSíndrome de Marfan SEMINARIOLuis GonzálezAún no hay calificaciones
- Ejer Cici OsDocumento39 páginasEjer Cici OsNashaly PérezAún no hay calificaciones
- Clase Autosomica DominanteDocumento66 páginasClase Autosomica DominanteCarlos D. S. PeñarandaAún no hay calificaciones
- Herencia Autonómica DominanteDocumento29 páginasHerencia Autonómica DominanteKarla Paola Juarez Sosa 2-06Aún no hay calificaciones
- SFGHJKDocumento7 páginasSFGHJKNathanael MartinezAún no hay calificaciones
- Genética del síndrome de MarfanDocumento42 páginasGenética del síndrome de MarfanAna Quispe IbericoAún no hay calificaciones
- Síndrome Marfan causas síntomasDocumento2 páginasSíndrome Marfan causas síntomasIsabel AntAún no hay calificaciones
- 1817 5996 Rcur 22 03 E769Documento11 páginas1817 5996 Rcur 22 03 E769Mary Flor Flores TapiaAún no hay calificaciones
- 2.6 Monografía EQ6Documento10 páginas2.6 Monografía EQ6Leslie de leónAún no hay calificaciones
- Médico oftalmólogo con amplia experiencia en subespecialidadesDocumento116 páginasMédico oftalmólogo con amplia experiencia en subespecialidadesJosé Angel MezaAún no hay calificaciones
- Pruebas Genéticas - Marfan, Síndrome De, (Marfan Syndrome) - Gen FBN1. - IVAMIDocumento2 páginasPruebas Genéticas - Marfan, Síndrome De, (Marfan Syndrome) - Gen FBN1. - IVAMILaura HordasAún no hay calificaciones
- Herencia MendelianaDocumento6 páginasHerencia MendelianayanerysAún no hay calificaciones
- Patologías GenéticasDocumento9 páginasPatologías GenéticasDaniela pAún no hay calificaciones
- Síndrome de Treacher-CollinsDocumento10 páginasSíndrome de Treacher-CollinsMarco Morante VegaAún no hay calificaciones
- Cromosoma 17 humanoDocumento16 páginasCromosoma 17 humanoIsidora Montalva MoragaAún no hay calificaciones
- 7ma. CLASE TEORICA DE PATOLOGIA GENERALDocumento61 páginas7ma. CLASE TEORICA DE PATOLOGIA GENERALΝτιέγκοAún no hay calificaciones
- Sindrome de Marfan 3Documento13 páginasSindrome de Marfan 3jhonparck394Aún no hay calificaciones
- Marfan, Alopecia Androgenica y Osteogenesis ImperfectaDocumento7 páginasMarfan, Alopecia Androgenica y Osteogenesis ImperfectaAndrea FerrazAún no hay calificaciones
- Síndrome de Marfán: A Propósito de Dos Casos Marfan Syndrome: Regarding Two CasesDocumento11 páginasSíndrome de Marfán: A Propósito de Dos Casos Marfan Syndrome: Regarding Two CasesLuz Adriana Castaño SuárezAún no hay calificaciones
- Defectos Congénitos-2021 PDFDocumento63 páginasDefectos Congénitos-2021 PDFMaria Fernanda Trejo TovarAún no hay calificaciones
- Sindrome de Marfan 1.1Documento5 páginasSindrome de Marfan 1.1Wilber SarmientoAún no hay calificaciones
- AcondroplasiaDocumento12 páginasAcondroplasiasamuel puentesAún no hay calificaciones
- CSDocumento19 páginasCSSamantha LopezAún no hay calificaciones
- Sindrome de MatDocumento6 páginasSindrome de MatlaportecarmenAún no hay calificaciones
- Sindrome de MarfanDocumento3 páginasSindrome de MarfanKARLA LISSETH BRAVO CEREZOAún no hay calificaciones
- Exposicion Sindrome de MarfanDocumento7 páginasExposicion Sindrome de MarfanMecos Crzp0% (1)
- Síndrome de MarfanDocumento4 páginasSíndrome de MarfanLaura HordasAún no hay calificaciones
- Trastornos de La MorfogénesisDocumento5 páginasTrastornos de La MorfogénesisMariana RamirezAún no hay calificaciones
- Abraham Lincoln y Sindrome MarfanDocumento7 páginasAbraham Lincoln y Sindrome MarfanRocio Yasminy Mamani YucraAún no hay calificaciones
- Síndrome de Marfán Ensayo 101Documento7 páginasSíndrome de Marfán Ensayo 101palomaAún no hay calificaciones
- S X Velo Cardio FacialDocumento30 páginasS X Velo Cardio FacialEleyra Llanos Parra100% (1)
- Síndrome de MarfanDocumento21 páginasSíndrome de MarfanJannete RamirezAún no hay calificaciones
- Síndrome de Goldenhar.Documento4 páginasSíndrome de Goldenhar.Natalia Vázquez EspírituAún no hay calificaciones
- Síndrome de Marfan: beneficios deportivosDocumento19 páginasSíndrome de Marfan: beneficios deportivosAnonymous vzLOUsAún no hay calificaciones
- EnfermedadesDocumento2 páginasEnfermedadesMarisol SerapioAún no hay calificaciones
- Embrilogia Sindrome ApertDocumento20 páginasEmbrilogia Sindrome ApertJuan PortillaAún no hay calificaciones
- Patrones Hereditarios Autosomica Dominante y RecesivaDocumento10 páginasPatrones Hereditarios Autosomica Dominante y RecesivaKriss León SanchezAún no hay calificaciones
- Sindrome de MarfanDocumento12 páginasSindrome de MarfanStefanny Soria RoblesAún no hay calificaciones
- Presentación (Creada Desde TuDocumento18 páginasPresentación (Creada Desde Tusindy garciaAún no hay calificaciones
- 2 Síndrome MarfanDocumento43 páginas2 Síndrome MarfanMaría Rosa MartínezAún no hay calificaciones
- Diagnosticos PLACE UPPDocumento4 páginasDiagnosticos PLACE UPPNicol RiveraAún no hay calificaciones
- Anexos 1,2 y 3 PaeDocumento11 páginasAnexos 1,2 y 3 PaeNicol RiveraAún no hay calificaciones
- Unidad 9. Farmacología Del Sistema EndocrinoDocumento16 páginasUnidad 9. Farmacología Del Sistema EndocrinoNicol RiveraAún no hay calificaciones
- Cuadro Holones de La SexualidadDocumento2 páginasCuadro Holones de La SexualidadNicol RiveraAún no hay calificaciones
- Avance Place ColelitiasisDocumento12 páginasAvance Place ColelitiasisNicol RiveraAún no hay calificaciones
- Sindrome de Marfan PresentacionDocumento12 páginasSindrome de Marfan PresentacionNicol RiveraAún no hay calificaciones
- EnsayoPoderdelaspalabras PDFDocumento5 páginasEnsayoPoderdelaspalabras PDFNicol RiveraAún no hay calificaciones
- Fichero de Farmacologia Parte 1 PDFDocumento158 páginasFichero de Farmacologia Parte 1 PDFNicol RiveraAún no hay calificaciones
- Historia de La EcologíaDocumento4 páginasHistoria de La EcologíaNicol RiveraAún no hay calificaciones
- El Noviazgo en La AdolecenciaDocumento1 páginaEl Noviazgo en La AdolecenciaNicol Rivera0% (1)
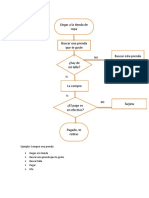
- Diagrama de FlujoDocumento2 páginasDiagrama de FlujoNicol RiveraAún no hay calificaciones
- Ensayo Comunicacón y ExpresiónDocumento6 páginasEnsayo Comunicacón y ExpresiónNicol RiveraAún no hay calificaciones
- Áreas Naturales ProtegidasDocumento3 páginasÁreas Naturales ProtegidasNicol RiveraAún no hay calificaciones
- COLLAGEDocumento2 páginasCOLLAGENicol RiveraAún no hay calificaciones
- Derecho LaboralDocumento1 páginaDerecho LaboralNicol RiveraAún no hay calificaciones
- Derecho PublicoDocumento1 páginaDerecho PublicoNicol RiveraAún no hay calificaciones
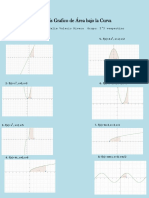
- Análisis Grafico de Área Bajo La CurvaDocumento1 páginaAnálisis Grafico de Área Bajo La CurvaNicol RiveraAún no hay calificaciones
- Conferencia - SaludDocumento1 páginaConferencia - SaludNicol RiveraAún no hay calificaciones
- Derecho Agrario-3°3vespDocumento1 páginaDerecho Agrario-3°3vespNicol RiveraAún no hay calificaciones
- Áreas Naturales ProtegidasDocumento3 páginasÁreas Naturales ProtegidasNicol RiveraAún no hay calificaciones
- Variadores de FrecuenciaDocumento2 páginasVariadores de FrecuenciaAriel ChicaizaAún no hay calificaciones
- La sucesión intestada en Roma: orden de herederos y reformasDocumento5 páginasLa sucesión intestada en Roma: orden de herederos y reformasJae Lita CarmenAún no hay calificaciones
- Cronograma de Control Concurrente AdministraciónDocumento1 páginaCronograma de Control Concurrente AdministraciónMarthaAún no hay calificaciones
- Cuaderno de Costos de Parcelas de Frijol o Ma - Z PDFDocumento21 páginasCuaderno de Costos de Parcelas de Frijol o Ma - Z PDFCristian Andres Gonzalez AlarconAún no hay calificaciones
- Clase 4Documento7 páginasClase 4Alejandro GodoyAún no hay calificaciones
- Unidad 3Documento9 páginasUnidad 3Luis NarvaezAún no hay calificaciones
- Periodoncia 1Documento7 páginasPeriodoncia 1Nathaly NathysaAún no hay calificaciones
- Cuadernillo Pendientes 3 ESO2 Evaluacion 2014Documento13 páginasCuadernillo Pendientes 3 ESO2 Evaluacion 2014Lucia Borràs PruñonosaAún no hay calificaciones
- Ficha Tecnica Cloro Al 91%Documento1 páginaFicha Tecnica Cloro Al 91%Luis GaviriaAún no hay calificaciones
- Bibliografia ATLAS DE AMERICA LATINA Y EL CARIBE PDFDocumento45 páginasBibliografia ATLAS DE AMERICA LATINA Y EL CARIBE PDFMicaela GarciaAún no hay calificaciones
- Introducción Al DerechoDocumento8 páginasIntroducción Al DerechoMárgara MartínezAún no hay calificaciones
- Cartilla Uno Gana Uno Mod II Edic2 VERDE PDFDocumento117 páginasCartilla Uno Gana Uno Mod II Edic2 VERDE PDFMARIO RENE GOMEZAún no hay calificaciones
- LMR Practica1Documento9 páginasLMR Practica1Liliana MontoyaAún no hay calificaciones
- Perfil BIPDocumento1 páginaPerfil BIPDaniela Sánchez AcostaAún no hay calificaciones
- Compra contra ConvenioDocumento21 páginasCompra contra Convenioboris fuentes riveraAún no hay calificaciones
- Guia Practica Del ISLRDocumento15 páginasGuia Practica Del ISLRElizabeth Cristina Santander Corrales91% (11)
- Triptico de InformeDocumento2 páginasTriptico de Informegrupoaepu100% (6)
- Pan Transformado en El Cuerpo de CristoDocumento10 páginasPan Transformado en El Cuerpo de CristowillymartinezsanchezAún no hay calificaciones
- Juventud Sin ValoresDocumento2 páginasJuventud Sin ValoresNiny Johanna Sandoval BuitragoAún no hay calificaciones
- Como Tener Cibersexualidad. Infidelidad Sexo Noviazgo y Casos Clínicos - Juan Antonio Guerrero CañongoDocumento122 páginasComo Tener Cibersexualidad. Infidelidad Sexo Noviazgo y Casos Clínicos - Juan Antonio Guerrero CañongoMartin Alejandro Menecier StimlerAún no hay calificaciones
- 9786079356149Documento12 páginas9786079356149Stefanía VieiraAún no hay calificaciones
- Entrenamiento 2023 05 17Documento3 páginasEntrenamiento 2023 05 17wallstreet cataAún no hay calificaciones
- Proyecto Aji DulceDocumento23 páginasProyecto Aji DulceLuiggi Ron100% (1)
- SALUD MENTAL CONDUCTA HUMANA EJERCICIODocumento14 páginasSALUD MENTAL CONDUCTA HUMANA EJERCICIONitram JonsAún no hay calificaciones
- Evaluación Derechos FundamentalesDocumento4 páginasEvaluación Derechos FundamentalesConstanzaAún no hay calificaciones
- Copia de Exse Vesm Sem03 Área C Con ClavesDocumento9 páginasCopia de Exse Vesm Sem03 Área C Con ClavesDiego CaparachinAún no hay calificaciones
- Comparativo Liberalismo y MarxismoDocumento2 páginasComparativo Liberalismo y MarxismoMaria Fernanda SalazarAún no hay calificaciones
- Tarea No. 6-Ensayo Lógica JurídicaDocumento2 páginasTarea No. 6-Ensayo Lógica JurídicaGabriela Rodas de LeónAún no hay calificaciones
- Caso Demanda de AlimentosDocumento2 páginasCaso Demanda de AlimentosActivoz promotora inmobiliariaAún no hay calificaciones
- El Financiero 13-06-23Documento36 páginasEl Financiero 13-06-23Open Course Ware MéxicoAún no hay calificaciones
- TDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosDe EverandTDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosCalificación: 4 de 5 estrellas4/5 (8)
- Terapia cognitiva: Conceptos básicos y profundizaciónDe EverandTerapia cognitiva: Conceptos básicos y profundizaciónCalificación: 5 de 5 estrellas5/5 (1)
- Trauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesDe EverandTrauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesCalificación: 1 de 5 estrellas1/5 (1)
- Psiconeuroinmunología para la práctica clínicaDe EverandPsiconeuroinmunología para la práctica clínicaCalificación: 5 de 5 estrellas5/5 (4)
- Fisiopatología de las enfermedades cardiovascularesDe EverandFisiopatología de las enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- El autismo: Reflexiones y pautas para comprenderlo y abordarloDe EverandEl autismo: Reflexiones y pautas para comprenderlo y abordarloCalificación: 4 de 5 estrellas4/5 (7)
- El libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)De EverandEl libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)Calificación: 3 de 5 estrellas3/5 (2)
- Ansiedad infantil. Los trastornos explicados a los padresDe EverandAnsiedad infantil. Los trastornos explicados a los padresCalificación: 4.5 de 5 estrellas4.5/5 (25)
- Prescripción de ejercico físico para la saludDe EverandPrescripción de ejercico físico para la saludCalificación: 5 de 5 estrellas5/5 (1)
- Psicoterapia breve con niños y adolescentesDe EverandPsicoterapia breve con niños y adolescentesCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Plan De Dieta Para La Resistencia A La Insulina & Sistema Inmunológico En EspañolDe EverandPlan De Dieta Para La Resistencia A La Insulina & Sistema Inmunológico En EspañolCalificación: 3.5 de 5 estrellas3.5/5 (2)
- Póngase En Forma Sin Salir De Su CasaDe EverandPóngase En Forma Sin Salir De Su CasaCalificación: 4.5 de 5 estrellas4.5/5 (4)
- Puntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)De EverandPuntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)Calificación: 4.5 de 5 estrellas4.5/5 (23)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)
- La revolución de la tiroides y las glándulas suprarrenales: Un programa detallado para equilibrar tu metabolismo, cuidar tus hormonas y mejorar tu estado de ánimoDe EverandLa revolución de la tiroides y las glándulas suprarrenales: Un programa detallado para equilibrar tu metabolismo, cuidar tus hormonas y mejorar tu estado de ánimoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Dieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasDe EverandDieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasCalificación: 4 de 5 estrellas4/5 (9)
- El código de la obesidad: Descifrando los secretos de la pérdida de pesoDe EverandEl código de la obesidad: Descifrando los secretos de la pérdida de pesoCalificación: 4.5 de 5 estrellas4.5/5 (51)
- Kinesiotaping: Pruebas musculares y aplicaciones de taping (Color)De EverandKinesiotaping: Pruebas musculares y aplicaciones de taping (Color)Calificación: 5 de 5 estrellas5/5 (4)