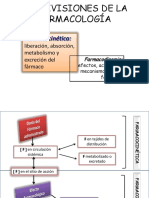
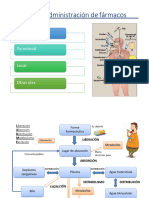
También podría gustarte
- Fármacología básica para el odontólogo: Conocimientos básicos odontológicos, #1De EverandFármacología básica para el odontólogo: Conocimientos básicos odontológicos, #1Calificación: 5 de 5 estrellas5/5 (4)
- Tecnicas Basicas de Enfermeria Medico Quirurgica Esa-114Documento162 páginasTecnicas Basicas de Enfermeria Medico Quirurgica Esa-114Vivian Sepulveda74% (19)
- FARMACOCINÉTICA - AbsorciónDocumento47 páginasFARMACOCINÉTICA - AbsorciónEmara MendezAún no hay calificaciones
- FarmacocinéticaDocumento53 páginasFarmacocinéticaSilvana Coraima Palacios Lavado100% (3)
- Expo S1-Fármaco Sem - Grupo 26-1Documento30 páginasExpo S1-Fármaco Sem - Grupo 26-1Lucía SilvaAún no hay calificaciones
- Requisitos para La Operación de FarmaciasDocumento36 páginasRequisitos para La Operación de Farmaciasfaserh100% (4)
- TEMA 3 - Bases Farmacocineticas. Absorcion y DistribucionDocumento40 páginasTEMA 3 - Bases Farmacocineticas. Absorcion y DistribucionLuis VelaAún no hay calificaciones
- Farmacocinetica - FarmacodinamiaDocumento85 páginasFarmacocinetica - FarmacodinamiaAndrea Camila Ramos Araujo100% (1)
- Farmacocinética y FarmacodinamiaDocumento91 páginasFarmacocinética y FarmacodinamiaCamilaCMartinezH100% (3)
- Farmacocinética y Farmacodinamia PDFDocumento83 páginasFarmacocinética y Farmacodinamia PDFsofia MogollonAún no hay calificaciones
- Clase 4 - FarmacodinamiaDocumento40 páginasClase 4 - FarmacodinamiaDeysi Yhulit CARHUARICRA ALANIAAún no hay calificaciones
- Farmacologia General (Azael)Documento76 páginasFarmacologia General (Azael)maryAún no hay calificaciones
- Farmacocinética DrSalasDocumento98 páginasFarmacocinética DrSalasCesar Del Aguila MattaAún no hay calificaciones
- Farmacocinética y FarmacodinamiaDocumento86 páginasFarmacocinética y FarmacodinamiamonylindaAún no hay calificaciones
- Principales Conceptos De: FarmacocinéticaDocumento23 páginasPrincipales Conceptos De: FarmacocinéticaMarioCraft 15Aún no hay calificaciones
- Fundamentos de FarmacologiaDocumento56 páginasFundamentos de FarmacologiamauroAún no hay calificaciones
- Clase Farmacologia General WebDocumento92 páginasClase Farmacologia General WebAgrotienda VasquezAún no hay calificaciones
- Fco Cinetica MetabolismoDocumento24 páginasFco Cinetica Metabolismovalentina perezAún no hay calificaciones
- FARMACOCINÉTICADocumento66 páginasFARMACOCINÉTICAMECHISSTAún no hay calificaciones
- Diapo Clase 2Documento23 páginasDiapo Clase 2Anna KareninaAún no hay calificaciones
- Farmacocinetica Karen2.022Documento27 páginasFarmacocinetica Karen2.022Alejandra ChaconAún no hay calificaciones
- CLASE 3 Farmacocinética I-1Documento30 páginasCLASE 3 Farmacocinética I-1LEO100% (1)
- 003 Farmacocinetica para ImprentaDocumento25 páginas003 Farmacocinetica para ImprentaJulio Cesar GutierrezAún no hay calificaciones
- Calculo y Dilucion de MedicamentosDocumento39 páginasCalculo y Dilucion de MedicamentosKarla HernándezAún no hay calificaciones
- Copia de Farmacocinetica 2023Documento55 páginasCopia de Farmacocinetica 2023Giovanni Jose Brizuela MaestreAún no hay calificaciones
- Modulo 2 FarmacocinéticaDocumento19 páginasModulo 2 FarmacocinéticaKarkeyAún no hay calificaciones
- 1 Bases de La FC ClínicaDocumento70 páginas1 Bases de La FC ClínicaMONTOYA CABANILLAS RAINIER IVANAún no hay calificaciones
- Farmaco Cinética y FarmacodinámicaDocumento38 páginasFarmaco Cinética y FarmacodinámicaLiza Melany TolentinoAún no hay calificaciones
- FARMACOCINÉTICADocumento241 páginasFARMACOCINÉTICAArturo MendozaAún no hay calificaciones
- 1er Trabajo. FarmacocinéticaDocumento30 páginas1er Trabajo. FarmacocinéticabrigetteAún no hay calificaciones
- Teoría 2. Farmacología-Unt-Farmacia y BioquimicaDocumento157 páginasTeoría 2. Farmacología-Unt-Farmacia y BioquimicaIsmael HuertasAún no hay calificaciones
- Clase 2 Farmacologia 2S 2020Documento29 páginasClase 2 Farmacologia 2S 2020Cha GamaAún no hay calificaciones
- Introduccion - Generalidades, FarmacologiaDocumento108 páginasIntroduccion - Generalidades, FarmacologialouisegantierAún no hay calificaciones
- Apuntes Farmacologia AlejandorJavierDocumento29 páginasApuntes Farmacologia AlejandorJaviersamanthaperez100% (2)
- Farmacologia IDocumento68 páginasFarmacologia IDaniel TrejoAún no hay calificaciones
- Clase 4 Definicióndelos Diferentes Términos Aplicados en FarmacoterapiaDocumento16 páginasClase 4 Definicióndelos Diferentes Términos Aplicados en FarmacoterapiaKarina MoralesAún no hay calificaciones
- Gallardo Itzamaná Resumen Temas 1.1 A 1.2Documento3 páginasGallardo Itzamaná Resumen Temas 1.1 A 1.2bumper.horizonAún no hay calificaciones
- 2 - Farmacocinetica y Farmaco DinamiaDocumento61 páginas2 - Farmacocinetica y Farmaco DinamiaSantiago OviedoAún no hay calificaciones
- Teoria 3Documento33 páginasTeoria 3HECTOR HEKERIN LUCAS TELLOAún no hay calificaciones
- 2° Mecanismo de Tranportes y AbsorcionDocumento43 páginas2° Mecanismo de Tranportes y AbsorcionluzAún no hay calificaciones
- FARMACOCINÉTICADocumento34 páginasFARMACOCINÉTICAPatrick Castillo Salazar100% (1)
- Tema 3 - Principios Básicos de FarmacocinéticaDocumento18 páginasTema 3 - Principios Básicos de FarmacocinéticaGuadalupe RoderoAún no hay calificaciones
- Farmacocinetica y FarmacodinamiaDocumento89 páginasFarmacocinetica y Farmacodinamiaana ramos martinezAún no hay calificaciones
- 1819 BfyFc Intr.Documento38 páginas1819 BfyFc Intr.Efrain PuchetaAún no hay calificaciones
- Farmacocinetica y FarmacodinamiaDocumento41 páginasFarmacocinetica y Farmacodinamiabetoasd95Aún no hay calificaciones
- Farmacología Quiz 2 Primer Corte-1Documento14 páginasFarmacología Quiz 2 Primer Corte-1liseth obandoAún no hay calificaciones
- Farmacocinetica y Farmacodinamia 2Documento40 páginasFarmacocinetica y Farmacodinamia 2Micaela Munayco SilvaAún no hay calificaciones
- FarmacocineticaDocumento36 páginasFarmacocineticaDulce LealAún no hay calificaciones
- Guía de Aprendizaje UABP 2Documento3 páginasGuía de Aprendizaje UABP 2Martín PanicoAún no hay calificaciones
- Teoricos y Talleres 2023Documento1117 páginasTeoricos y Talleres 2023Aldana Jazmin FrancoAún no hay calificaciones
- FarmacologíaDocumento41 páginasFarmacologíaAnggy PantaAún no hay calificaciones
- 2 FarmacocineticaDocumento33 páginas2 FarmacocineticaCarlos RiquelmeAún no hay calificaciones
- FarmacocinticaDocumento23 páginasFarmacocinticaWILDER NUÑUVERO ARCOSAún no hay calificaciones
- Farmacocinética y FarmacodinamiaDocumento80 páginasFarmacocinética y FarmacodinamiaAxel Castro100% (5)
- Fco 1 ParteDocumento99 páginasFco 1 ParteMaria Concepción Chura Barón100% (1)
- T1. Parte ADocumento18 páginasT1. Parte ACristina MartinezAún no hay calificaciones
- Procesos Del Farmaco en El Organismo PDFDocumento41 páginasProcesos Del Farmaco en El Organismo PDFpaucar sulla janethAún no hay calificaciones
- Toxico Clase Cuatro 01 SeptDocumento29 páginasToxico Clase Cuatro 01 SeptRhebeca MurbachAún no hay calificaciones
- Metabolismo del sistema digestivo, del hígado, de la vesícula y de las vías biliares: En condiciones de salud y en las enfermedadesDe EverandMetabolismo del sistema digestivo, del hígado, de la vesícula y de las vías biliares: En condiciones de salud y en las enfermedadesAún no hay calificaciones
- El cannabis medicinal y su dosificaciónDe EverandEl cannabis medicinal y su dosificaciónCalificación: 3 de 5 estrellas3/5 (2)
- Gans and Kligman (2000) Resurgimiento Del Retinol Tópico en La DermatologíaDocumento13 páginasGans and Kligman (2000) Resurgimiento Del Retinol Tópico en La DermatologíaPér Hur ViviAún no hay calificaciones
- Perfil Profesional Del EducadorDocumento10 páginasPerfil Profesional Del Educadorirene191Aún no hay calificaciones
- Cuadro Matriz de RiesgosDocumento24 páginasCuadro Matriz de RiesgosMarta Cecilia Acosta CalleAún no hay calificaciones
- C AsmDocumento4 páginasC AsmJavier Fernando Guerrero DuránAún no hay calificaciones
- Medicamentos AntihispaminicosDocumento6 páginasMedicamentos AntihispaminicosJuany MadrigalAún no hay calificaciones
- PDF 20231117 090706 0000Documento10 páginasPDF 20231117 090706 0000anasuzzet28Aún no hay calificaciones
- Evidencia 3 Régimen LegalDocumento4 páginasEvidencia 3 Régimen LegalYolandaAún no hay calificaciones
- Contribución de La Enfermería en La Farmacovigilancia HospitalariaDocumento63 páginasContribución de La Enfermería en La Farmacovigilancia HospitalariaJuan Carlos Miranda RodriguezAún no hay calificaciones
- Práctica 9 - Tecnología FarmacéuticaDocumento7 páginasPráctica 9 - Tecnología FarmacéuticaEmiliano RodriguezAún no hay calificaciones
- DOCUMENTO HomeopáticosDocumento13 páginasDOCUMENTO HomeopáticosKamilo ÁlvarezAún no hay calificaciones
- Gases MedicinalesDocumento27 páginasGases MedicinalesLiliana SanchezAún no hay calificaciones
- Iridologia Simplificada CorregidaDocumento67 páginasIridologia Simplificada CorregidaJaviernaturopata100% (7)
- Vías de Administración de MedicamentosDocumento42 páginasVías de Administración de MedicamentosAlejandroAún no hay calificaciones
- Manual de Manejo de Medicamentos de Control EspecialDocumento19 páginasManual de Manejo de Medicamentos de Control EspecialJenifer CaceresAún no hay calificaciones
- Cuidados IntensivosDocumento50 páginasCuidados Intensivosavargas_423700100% (1)
- TP DeontoDocumento4 páginasTP DeontoSabri BlueAún no hay calificaciones
- Manual de Buenas Prácticas FarmacéuticasDocumento5 páginasManual de Buenas Prácticas Farmacéuticasaspire726Aún no hay calificaciones
- CASO CLINICO Nanda Noc NicDocumento11 páginasCASO CLINICO Nanda Noc NicDayanna RodriguezAún no hay calificaciones
- Tocto Chavesta YarumiDocumento8 páginasTocto Chavesta YarumiYarumi Tocto ChavestaAún no hay calificaciones
- Proyecto Resolución UnidosisDocumento4 páginasProyecto Resolución UnidosisLis EnsalanderAún no hay calificaciones
- Formas Farmacéuticas Líquidas y SemisólidasDocumento4 páginasFormas Farmacéuticas Líquidas y SemisólidasSANDRO JESUS YUPANQUI100% (1)
- Guia de Enfermeria OstomiaDocumento41 páginasGuia de Enfermeria OstomiaPaola RodriguezAún no hay calificaciones
- Terceros Autorizados 2014Documento56 páginasTerceros Autorizados 2014Alejandra MedinaAún no hay calificaciones
- Estrategias de ColaboraciónDocumento1 páginaEstrategias de ColaboraciónXimenaAún no hay calificaciones
- Charla de 5 Min (27-01-2015)Documento45 páginasCharla de 5 Min (27-01-2015)JuanCervantesMiovichAún no hay calificaciones
- Atencion GeriatricaDocumento5 páginasAtencion GeriatricaLider Rojas CanoAún no hay calificaciones
- Terminologia ResumenDocumento45 páginasTerminologia ResumenAngie Ana Sofia Morales MolinerosAún no hay calificaciones
- Silabo FARMACIA QUIMICA IIDocumento5 páginasSilabo FARMACIA QUIMICA IIManuel NinahuancaAún no hay calificaciones