También podría gustarte
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleAún no hay calificaciones
- FISIOLOGIADocumento33 páginasFISIOLOGIALuis Octavio VillanuevaAún no hay calificaciones
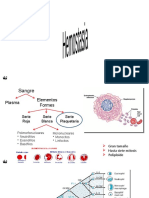
- Hemostasia: Mecanismos y factores de coagulación sanguíneaDocumento38 páginasHemostasia: Mecanismos y factores de coagulación sanguíneaJosé kreidy Herrera mena100% (2)
- Hemostasia Primaria 2017Documento80 páginasHemostasia Primaria 2017Josué García FloresAún no hay calificaciones
- Patologia Semana 9 y 10 ResumenDocumento13 páginasPatologia Semana 9 y 10 ResumenRita MazaAún no hay calificaciones
- Teoria 3Documento75 páginasTeoria 3Fatima AlvarezAún no hay calificaciones
- Hemostasia 130217220940 Phpapp02Documento50 páginasHemostasia 130217220940 Phpapp02Dominic Shelby CervantesAún no hay calificaciones
- CoagulaciónDocumento29 páginasCoagulaciónRocio FabianAún no hay calificaciones
- La HemostasiaDocumento8 páginasLa HemostasiaJOSE AAún no hay calificaciones
- HemostasiaDocumento52 páginasHemostasiaGeanella PadillaAún no hay calificaciones
- Plaquetas - Hemostasia - F. CoagulacionDocumento30 páginasPlaquetas - Hemostasia - F. CoagulacionBOTY HerAún no hay calificaciones
- CoagulaciónDocumento39 páginasCoagulaciónolgaAún no hay calificaciones
- 21 HemostasiaDocumento28 páginas21 HemostasiaFernando SenaAún no hay calificaciones
- Coagulación: Proceso de formación de fibrinaDocumento32 páginasCoagulación: Proceso de formación de fibrinaJosué García Flores50% (2)
- Coagulación SanguíneaDocumento52 páginasCoagulación SanguíneaPablo LucioAún no hay calificaciones
- HemostasiaDocumento27 páginasHemostasiaHernández Barragán YaritzaAún no hay calificaciones
- Hemostasia - 1Documento32 páginasHemostasia - 1wilmerAún no hay calificaciones
- Hemostasia y CoagulacionDocumento33 páginasHemostasia y CoagulacionElvis Yon GutierrezAún no hay calificaciones
- Cascada de La CoagulaciónDocumento37 páginasCascada de La CoagulacióndavidAún no hay calificaciones
- Hemostasia: proceso complejo para prevenir pérdida sangreDocumento32 páginasHemostasia: proceso complejo para prevenir pérdida sangrewilmerAún no hay calificaciones
- Anticoagulantes y AntiagregantesDocumento4 páginasAnticoagulantes y AntiagregantesHannia Montserrat Yañez VazquezAún no hay calificaciones
- Sindrome Hemorragico-PediatríaDocumento50 páginasSindrome Hemorragico-PediatríaNATALIA SALAMANCA PEÑAAún no hay calificaciones
- Anticoagulantes Orales y FibrinolisisDocumento22 páginasAnticoagulantes Orales y FibrinolisisMonsePuenteAún no hay calificaciones
- Fisiologia HemostasiaDocumento44 páginasFisiologia HemostasiaIsmael MedinaAún no hay calificaciones
- 05 Fisiologia de Las Plaquetas y Factores de CoagulaciónDocumento29 páginas05 Fisiologia de Las Plaquetas y Factores de Coagulaciónjavier ochoaAún no hay calificaciones
- Capitulo. 37 Hemostasia y Coagulación SanguineaDocumento24 páginasCapitulo. 37 Hemostasia y Coagulación SanguineaHelmy Mendez Mejia100% (2)
- Apunte de Quimica de Proteinas Plasmaticas UABP2Documento69 páginasApunte de Quimica de Proteinas Plasmaticas UABP2Jimena RenieroAún no hay calificaciones
- Coagulación y FibrinólisisDocumento31 páginasCoagulación y FibrinólisisRodrigo MayorgaAún no hay calificaciones
- Hemostasia y CoagulaciónDocumento14 páginasHemostasia y CoagulaciónAbraham LopezAún no hay calificaciones
- Hemostasia, Coagulacion y PlaquetasDocumento47 páginasHemostasia, Coagulacion y PlaquetasYHUMY CARRION UTANIAún no hay calificaciones
- Hemostasia y Trombosis AnatopatoDocumento10 páginasHemostasia y Trombosis Anatopatoss501100% (1)
- Hemostasia CLASEDocumento31 páginasHemostasia CLASEkennydi Benites GalindosAún no hay calificaciones
- Teoria HemostasiaDocumento41 páginasTeoria HemostasiaErik J. Roshan Flores100% (1)
- Trastornos HemodinamicosDocumento75 páginasTrastornos HemodinamicosBrandon AlanisAún no hay calificaciones
- Hemostasian 1Documento28 páginasHemostasian 1micaAún no hay calificaciones
- Hemostasia PrimariaDocumento32 páginasHemostasia PrimariaJuan Sebastian Ardila50% (2)
- Hemostasia: Adhesión y agregación plaquetarDocumento56 páginasHemostasia: Adhesión y agregación plaquetarJhonny IbañezAún no hay calificaciones
- 3 HemostasiaDocumento37 páginas3 HemostasiaTøfuAún no hay calificaciones
- HEMOSTASIA Y COAGULACIONDocumento30 páginasHEMOSTASIA Y COAGULACIONEsme p.Aún no hay calificaciones
- Cascada de CoagulacionDocumento17 páginasCascada de CoagulacionMari ChuAún no hay calificaciones
- SÍNDROME HEMORRAGÍPARODocumento76 páginasSÍNDROME HEMORRAGÍPAROJENNYAún no hay calificaciones
- Hemostasia Fundamentos 2012Documento38 páginasHemostasia Fundamentos 2012Christian RodriguezAún no hay calificaciones
- 6 Hemostasiaycoagulacin-110711200910-Phpapp01Documento54 páginas6 Hemostasiaycoagulacin-110711200910-Phpapp01Nelson BrunoAún no hay calificaciones
- Power Coagulacion y HemostasiaDocumento28 páginasPower Coagulacion y HemostasiaCinthya Melendez100% (1)
- Hemato II CompletaDocumento387 páginasHemato II CompletaMishellAún no hay calificaciones
- Semana 4Documento21 páginasSemana 4ANDREA DAYANH CRISTOBAL HINOSTROZAAún no hay calificaciones
- SANGRE Definitivo - PPTMDocumento33 páginasSANGRE Definitivo - PPTMSERGIO TOPETEAún no hay calificaciones
- Sistema hemostáticoDocumento58 páginasSistema hemostáticoSerafin Fernández100% (1)
- Fisiologia de La CoagulaciónDocumento32 páginasFisiologia de La CoagulaciónFrancoQuirozSanchez100% (1)
- Coagulacion Cascadas OKDocumento47 páginasCoagulacion Cascadas OKjoselinAún no hay calificaciones
- 3 - Fisiologia de La Sangre - Hemostasia - 2014-IDocumento46 páginas3 - Fisiologia de La Sangre - Hemostasia - 2014-IDharyll Chancasanampa NarvaezAún no hay calificaciones
- Fibrinogeno, Protrombina y Cascada de CoagulacionDocumento44 páginasFibrinogeno, Protrombina y Cascada de CoagulacionRobi GeltAún no hay calificaciones
- Factores de CoagulacionDocumento26 páginasFactores de CoagulacionlizisabellinarezpandAún no hay calificaciones
- Fisiología de La HemostasiaDocumento85 páginasFisiología de La HemostasiajosemaortizAún no hay calificaciones
- Recuento de PlaquetasDocumento21 páginasRecuento de PlaquetasALANAún no hay calificaciones
- Sistema de coagulación: procesos, pruebas y regulaciónDocumento26 páginasSistema de coagulación: procesos, pruebas y regulaciónJesúsLoya100% (1)
- Homeostasis coagulaciónDocumento3 páginasHomeostasis coagulaciónYesenia JimenezAún no hay calificaciones
- 17 Antiagregantes y AnticoagulantesDocumento102 páginas17 Antiagregantes y AnticoagulantesVANESSA RINCON RODRIGUEZAún no hay calificaciones
- 5 Pruebas HemostásicasDocumento2 páginas5 Pruebas HemostásicasEVELIN MICHEL DE LA CRUZ RUIZAún no hay calificaciones
- Monografía ILSI. Hidratación PDFDocumento39 páginasMonografía ILSI. Hidratación PDFnaudy carmona100% (1)
- Sindrome HemorragicoDocumento34 páginasSindrome HemorragicoBryan Flores SierraAún no hay calificaciones
- Hda ManagmentDocumento54 páginasHda ManagmentWeymar Poma LunaAún no hay calificaciones
- Toma de Muestra 01 1Documento13 páginasToma de Muestra 01 1Morelia Sofia Garcia OlarteAún no hay calificaciones
- Fisiologia AnimalDocumento9 páginasFisiologia AnimalJuan Camilo Yepes0% (1)
- HEMATOLOGÍADocumento36 páginasHEMATOLOGÍAJazmin AliciaAún no hay calificaciones
- ResultadoLaboratorio PDFDocumento3 páginasResultadoLaboratorio PDFamalia sanabriaAún no hay calificaciones
- Aparato CardiovascularDocumento59 páginasAparato CardiovascularAmerica MendozaAún no hay calificaciones
- Alg Lab Hep 29 Dic 2021Documento4 páginasAlg Lab Hep 29 Dic 2021Alex López GonzálezAún no hay calificaciones
- Compomat G5 OP 06 02 12 SW 01 01 ESDocumento138 páginasCompomat G5 OP 06 02 12 SW 01 01 ESwilliams luis arana floriano100% (1)
- Universidad Tecnologica de SantiagoDocumento9 páginasUniversidad Tecnologica de SantiagoJeferson TbAún no hay calificaciones
- La educación intercultural bilingüe y el desarrollo lingüísticoDocumento27 páginasLa educación intercultural bilingüe y el desarrollo lingüísticoAdriel ManzanoAún no hay calificaciones
- Parte I Seminario de Generalidades de La Medicina LegalDocumento57 páginasParte I Seminario de Generalidades de La Medicina Legalallylaura161297Aún no hay calificaciones
- Sistema linfático y sus funcionesDocumento2 páginasSistema linfático y sus funcionesAlejandro Huallanca ZamudioAún no hay calificaciones
- Unidad 3 - Tarea 5 - Componente PrácticoDocumento92 páginasUnidad 3 - Tarea 5 - Componente PrácticoJose Anderson CamposAún no hay calificaciones
- Tranfusion Sanguinea.1docxDocumento11 páginasTranfusion Sanguinea.1docxCarolina HuertasAún no hay calificaciones
- Microscopia en Los TejidosDocumento5 páginasMicroscopia en Los TejidosJose AcostaAún no hay calificaciones
- Enfermedades Que Afectan Al Sistema CirculatorioDocumento2 páginasEnfermedades Que Afectan Al Sistema CirculatorioValeria Fabiola Farias MarambioAún no hay calificaciones
- RAE Explicar El Origen Del Uso Del Laboratorio en La Medicina Moderna.Documento6 páginasRAE Explicar El Origen Del Uso Del Laboratorio en La Medicina Moderna.Laura Sofia Peña PerezAún no hay calificaciones
- Informe Lab. FisioDocumento15 páginasInforme Lab. FisioMariely HernándezAún no hay calificaciones
- Invitacion A CotizarDocumento3 páginasInvitacion A CotizarCrida EngoAún no hay calificaciones
- Taller en Clases Laboratorio ClinicoDocumento11 páginasTaller en Clases Laboratorio Cliniconicol michel diaz herreraAún no hay calificaciones
- Manual Acondicionamiento Fisico para BomberosDocumento175 páginasManual Acondicionamiento Fisico para Bomberosdiegomolina8394% (16)
- RepasoDocumento7 páginasRepasoOmar BarraganAún no hay calificaciones
- HEMOVIGILANCIADocumento59 páginasHEMOVIGILANCIAMaria Andrea OyolaAún no hay calificaciones
- Unidad IDocumento26 páginasUnidad IAlejandro LunaAún no hay calificaciones
- Practica n0. 2 Biología CelularDocumento26 páginasPractica n0. 2 Biología CelularOsneider Arroyo0% (1)
- Subrayado y EsquemaDocumento32 páginasSubrayado y Esquemamabel_cuencaAún no hay calificaciones
- B - 5° - S1 - Sistema Circulatorio ComparadoDocumento2 páginasB - 5° - S1 - Sistema Circulatorio ComparadojaimecruzqAún no hay calificaciones
- Fisiologia PulmonesDocumento28 páginasFisiologia PulmonesDayanara RodriguezAún no hay calificaciones