También podría gustarte
- Instrumentación y laboratorio. Manual de procedimientos básicosDe EverandInstrumentación y laboratorio. Manual de procedimientos básicosCalificación: 3 de 5 estrellas3/5 (1)
- Uso micropipetas laboratorioDocumento6 páginasUso micropipetas laboratorioMally Yasmin Torres SanchezAún no hay calificaciones
- Manual de practicas de laboratorio de MicrobiologíaDe EverandManual de practicas de laboratorio de MicrobiologíaCalificación: 4 de 5 estrellas4/5 (7)
- Plantilla para PNTDocumento8 páginasPlantilla para PNTJM A MAún no hay calificaciones
- Mediciones y métodos de uso común en el laboratorio de QuímicaDe EverandMediciones y métodos de uso común en el laboratorio de QuímicaCalificación: 4.5 de 5 estrellas4.5/5 (3)
- Guia de Lab. Bio. Mol PDFDocumento47 páginasGuia de Lab. Bio. Mol PDFSteven Agredo SánchezAún no hay calificaciones
- USO DE MICRO PIPETA GENERALDocumento6 páginasUSO DE MICRO PIPETA GENERALvtoledoopimi100% (1)
- La Micropipeta.Documento9 páginasLa Micropipeta.Mally Yasmin Torres SanchezAún no hay calificaciones
- MicropipetasDocumento9 páginasMicropipetasKalethJAún no hay calificaciones
- 5 - Manual de Biología Molecular IIDocumento16 páginas5 - Manual de Biología Molecular IIIsmael GuerreroAún no hay calificaciones
- Manual de Laboratorio de Biologia Molecular TP 2021Documento40 páginasManual de Laboratorio de Biologia Molecular TP 2021Mabel CaicedoAún no hay calificaciones
- Uso de MicropipetasDocumento8 páginasUso de MicropipetasMaria Gala Saavedra UribeAún no hay calificaciones
- Manual LTM Version 2022Documento88 páginasManual LTM Version 2022Diego Ismael VargasAún no hay calificaciones
- Uso de micropipetas y esterilización en biología molecularDocumento3 páginasUso de micropipetas y esterilización en biología molecularManuel CanoAún no hay calificaciones
- Informe Micropipeta EispdmDocumento10 páginasInforme Micropipeta EispdmMiriam Leticia Sanabria AliagaAún no hay calificaciones
- Informe N°1 Micropipetas (MARJEORY BRISSETH CACHAY GONZALES)Documento7 páginasInforme N°1 Micropipetas (MARJEORY BRISSETH CACHAY GONZALES)Marjeory Brisseth Cachay GonzalesAún no hay calificaciones
- Informe PipetasDocumento5 páginasInforme PipetasoscarAún no hay calificaciones
- ACTIVIDAD - Uso de Material de LaboratorioDocumento8 páginasACTIVIDAD - Uso de Material de LaboratorioNatt MejaiAún no hay calificaciones
- Sem 14 Uso de MicropipetaDocumento9 páginasSem 14 Uso de MicropipetaJennifer Obregon LopezAún no hay calificaciones
- Uso micropipetas biología molecularDocumento5 páginasUso micropipetas biología molecularMayra Alejandra100% (6)
- Lab 1 Calibración de MicropipetasDocumento4 páginasLab 1 Calibración de Micropipetasabdiel0% (1)
- Manual Bioconversiones 2020 - LFL - 21BDocumento85 páginasManual Bioconversiones 2020 - LFL - 21Bjose juanAún no hay calificaciones
- Manual InmunoDocumento81 páginasManual InmunoYesenia GallegosAún no hay calificaciones
- Manual Biología Molecular 2019 PDFDocumento48 páginasManual Biología Molecular 2019 PDFJoseRuizMontañoAún no hay calificaciones
- Práctica 1. Uso de Las Micropipetas.Documento5 páginasPráctica 1. Uso de Las Micropipetas.ALAN JABDIEL FLORES ENCINASAún no hay calificaciones
- Técnicas de Laboratorio Asépticas Ransferencias de Volumen Con Pipetas Serológicas y Micropipetas PDFDocumento16 páginasTécnicas de Laboratorio Asépticas Ransferencias de Volumen Con Pipetas Serológicas y Micropipetas PDFDayana MerinoAún no hay calificaciones
- LA MICROPIPETA - InformeDocumento9 páginasLA MICROPIPETA - InformeMally Yasmin Torres SanchezAún no hay calificaciones
- Precisión micropipetas automáticasDocumento4 páginasPrecisión micropipetas automáticasmadeleissy PerezAún no hay calificaciones
- Micropipeta Trabajo FinalDocumento7 páginasMicropipeta Trabajo FinalDianaMejiaAlayoAún no hay calificaciones
- Lab. Biquimica Clinica Practicas 1,2,3,4Documento8 páginasLab. Biquimica Clinica Practicas 1,2,3,4Andrea MoralesAún no hay calificaciones
- Uso y manejo de micropipetasDocumento8 páginasUso y manejo de micropipetasHeidi YomaraAún no hay calificaciones
- P2 - Guia - Micropipetas y Microdiluciones INI Sem1Documento11 páginasP2 - Guia - Micropipetas y Microdiluciones INI Sem1Fernanda NavarroAún no hay calificaciones
- Manual Analisis BacterilogicosDocumento29 páginasManual Analisis BacterilogicosJose Bruno Apodaca FuentesAún no hay calificaciones
- Instrumentacion de LaboratorioDocumento25 páginasInstrumentacion de LaboratorioVictoria Castillo GomezAún no hay calificaciones
- Práctica 2 de Ecología "Pipeteo"Documento7 páginasPráctica 2 de Ecología "Pipeteo"Tabata SantanaAún no hay calificaciones
- Guia-El Uso de Micropipeta-BMDocumento7 páginasGuia-El Uso de Micropipeta-BMMiguel Angel Perez100% (2)
- Practica 1. Fundamentos Basicos de Bioseguridad IG.Documento6 páginasPractica 1. Fundamentos Basicos de Bioseguridad IG.Norma DiazAún no hay calificaciones
- Practica IntroductoriaDocumento4 páginasPractica IntroductoriaFernanda MartinezAún no hay calificaciones
- MICROPIPETASDocumento6 páginasMICROPIPETASValeria Ariaas AngelAún no hay calificaciones
- Practica 1 Micro LaboratorioDocumento7 páginasPractica 1 Micro LaboratorioNila MoreanoAún no hay calificaciones
- Manual InmunologíaDocumento81 páginasManual InmunologíaKevin smithAún no hay calificaciones
- Práctica de MicropipeteoDocumento10 páginasPráctica de MicropipeteoJuzif AltairAún no hay calificaciones
- MANUAL DE PRACTICAS Química.Documento64 páginasMANUAL DE PRACTICAS Química.Fer Aguilar ManriqueAún no hay calificaciones
- Verificación de micro pipetas fotométricaDocumento3 páginasVerificación de micro pipetas fotométricaGUADALUPEAún no hay calificaciones
- Practica 3 MicropipetasDocumento3 páginasPractica 3 Micropipetasoskr205 uliAún no hay calificaciones
- Examen General de Orina Fisico y QuimicoDocumento9 páginasExamen General de Orina Fisico y QuimicoJasmin RojasAún no hay calificaciones
- Fla 23 Guia Unifica Laboratori 1Documento2 páginasFla 23 Guia Unifica Laboratori 1kimberly katherine quiroga lealAún no hay calificaciones
- Guía AEP1Documento10 páginasGuía AEP1Iris RodriguezAún no hay calificaciones
- Gips 5 AnalisisDocumento7 páginasGips 5 AnalisisKATIA PADILLA GARCIAAún no hay calificaciones
- COMPILADO Enero 2018Documento85 páginasCOMPILADO Enero 2018Sofia GlezAún no hay calificaciones
- INFORME-CULTIVO-MICROALGAS ListoDocumento10 páginasINFORME-CULTIVO-MICROALGAS ListoIgnacio GuzmanAún no hay calificaciones
- Uso de Micropipetas BiomolDocumento5 páginasUso de Micropipetas BiomolEvelyn SalomónAún no hay calificaciones
- USO DE MICROPIPETASDocumento5 páginasUSO DE MICROPIPETASsaruausAún no hay calificaciones
- Práctica 5. Micropipetas CarlosDocumento2 páginasPráctica 5. Micropipetas CarlosAroa Gongora OrtizAún no hay calificaciones
- IntroducciónDocumento7 páginasIntroducciónalberto.am0104Aún no hay calificaciones
- Determinación de densidadDocumento9 páginasDeterminación de densidadnatalia perezAún no hay calificaciones
- Bioseguridad InfDocumento12 páginasBioseguridad InfLuciel HilaresAún no hay calificaciones
- Informe de Analisis Cualitativo Del Agua BQM (GR-1)Documento56 páginasInforme de Analisis Cualitativo Del Agua BQM (GR-1)Josué Marcos RamírezAún no hay calificaciones
- PracticaDocumento15 páginasPracticaNicanor JimenezAún no hay calificaciones
- Practica 1 Biologia MolecularDocumento4 páginasPractica 1 Biologia MolecularKarla Karina Rodriguez RoblesAún no hay calificaciones
- Papel de TGF-B en ARDocumento17 páginasPapel de TGF-B en ARAlejandro Pérez GonzálezAún no hay calificaciones
- 1613 Biologia V - 2022-2023Documento14 páginas1613 Biologia V - 2022-2023ANA FERNANDA AGUILAR SOLISAún no hay calificaciones
- Sección W43 - Plan 14 - Primer Post Test - Attempt ReviewDocumento5 páginasSección W43 - Plan 14 - Primer Post Test - Attempt Reviewenmanueltaverasp027Aún no hay calificaciones
- Acción y mecanismo de los estrógenosDocumento2 páginasAcción y mecanismo de los estrógenosPamela ItalAún no hay calificaciones
- Procariota Estructuras Mem, Microcorpatimientos, InclusionesDocumento11 páginasProcariota Estructuras Mem, Microcorpatimientos, InclusionesAngela HernandezAún no hay calificaciones
- Fisiologia Del Esfuerzo FisicoDocumento190 páginasFisiologia Del Esfuerzo FisicoedyAún no hay calificaciones
- Guia EXPRESION GÉNICADocumento5 páginasGuia EXPRESION GÉNICAFernanda SánchezAún no hay calificaciones
- Metabolismo de Los Carbohidratos y Su Relación BioenergéticaDocumento40 páginasMetabolismo de Los Carbohidratos y Su Relación BioenergéticaAnthony Xavier Tlv Ponce García100% (1)
- Ruta de Las Pentosas Fosfato PDFDocumento23 páginasRuta de Las Pentosas Fosfato PDFAlexis JenatzyAún no hay calificaciones
- Fisiología FuncionesDocumento7 páginasFisiología FuncionesMorejon Aguila Jose AlejandroAún no hay calificaciones
- Ciclo Celular 2020 PDFDocumento7 páginasCiclo Celular 2020 PDFleslie rivasAún no hay calificaciones
- Aa. y Peptidos Con Interes BiologicoDocumento16 páginasAa. y Peptidos Con Interes BiologicoBeatrízLeónAún no hay calificaciones
- Clasificación de BaltimoreDocumento1 páginaClasificación de BaltimoreSmirf FeimsAún no hay calificaciones
- Informe Proteinas JorgeDocumento20 páginasInforme Proteinas JorgeJ Mora GañanAún no hay calificaciones
- Estrategias CatalíticasDocumento20 páginasEstrategias CatalíticasjoselynAún no hay calificaciones
- FENOMENOS DE TRANSPORTE FinalDocumento9 páginasFENOMENOS DE TRANSPORTE FinalSilviaCarrascalAún no hay calificaciones
- Celula Eucariota y OrganelosDocumento19 páginasCelula Eucariota y Organelosandreina255910abAún no hay calificaciones
- 2020planificacion BiologíaDocumento8 páginas2020planificacion BiologíaEstefania De BattistaAún no hay calificaciones
- DIAPOSITIVAS - Cidos NucleicosDocumento37 páginasDIAPOSITIVAS - Cidos NucleicosLucero MendozaAún no hay calificaciones
- Biocel: Fundamentos célula saludDocumento5 páginasBiocel: Fundamentos célula saludJudyth Barrera GAún no hay calificaciones
- TP Citologia AnimalDocumento61 páginasTP Citologia AnimalGustavo A. SuarezAún no hay calificaciones
- BioquimicaDocumento126 páginasBioquimicaSousuke SagaraAún no hay calificaciones
- Farmacodinamica de Farmacos ReceptoresDocumento65 páginasFarmacodinamica de Farmacos ReceptoresLeny Chuquicaña Quispe100% (1)
- Caso 7, Semana 13-Regulación de La Expresión Génica 2Documento6 páginasCaso 7, Semana 13-Regulación de La Expresión Génica 2LAURA LEONOR VITELLA CASTROAún no hay calificaciones
- Biologia10° Y11°Documento4 páginasBiologia10° Y11°variedades llamadasAún no hay calificaciones
- Los LisomasDocumento1 páginaLos LisomasAna EspinolaAún no hay calificaciones
- Cardona - Los Aminoácidos. Estructura y Tipos.Documento7 páginasCardona - Los Aminoácidos. Estructura y Tipos.Gaby FloresAún no hay calificaciones
- Farmacocinetica y Farmacodinamia PDFDocumento77 páginasFarmacocinetica y Farmacodinamia PDFTAREAS JOTAAún no hay calificaciones
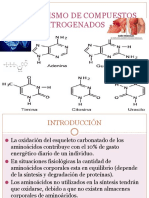
- Metabolismo de Compuestos NitrogenadosDocumento53 páginasMetabolismo de Compuestos NitrogenadosIsai GonzalezAún no hay calificaciones
- Metabolismo Proteínas-EstDocumento32 páginasMetabolismo Proteínas-Estgerson1contreras-1Aún no hay calificaciones
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- La revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaDe EverandLa revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaCalificación: 5 de 5 estrellas5/5 (200)
- Neurociencia para vencer la depresión: La esprial ascendenteDe EverandNeurociencia para vencer la depresión: La esprial ascendenteCalificación: 4.5 de 5 estrellas4.5/5 (10)
- Anatomía & 100 estiramientos Esenciales (Color): Técnicas, beneficios, precauciones, consejos, tablas de series, dolenciasDe EverandAnatomía & 100 estiramientos Esenciales (Color): Técnicas, beneficios, precauciones, consejos, tablas de series, dolenciasCalificación: 4.5 de 5 estrellas4.5/5 (21)
- Reconstrucción de dientes endodonciados: Pautas de actuación clínicaDe EverandReconstrucción de dientes endodonciados: Pautas de actuación clínicaCalificación: 5 de 5 estrellas5/5 (4)
- Fisiopatología de las enfermedades cardiovascularesDe EverandFisiopatología de las enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)
- Desastres y emergencias. Prevención, mitigación y preparaciónDe EverandDesastres y emergencias. Prevención, mitigación y preparaciónCalificación: 4 de 5 estrellas4/5 (5)
- Resumen de Pensar rápido pensar despacio de Daniel KahnemanDe EverandResumen de Pensar rápido pensar despacio de Daniel KahnemanCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Interpretación del ECG: Una Guía Práctica e Intuitiva para Aprender a Leer el ECG y Diagnosticar y Tratar ArritmiasDe EverandInterpretación del ECG: Una Guía Práctica e Intuitiva para Aprender a Leer el ECG y Diagnosticar y Tratar ArritmiasAún no hay calificaciones
- Disciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreDe EverandDisciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreCalificación: 5 de 5 estrellas5/5 (3)
- Batidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoDe EverandBatidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoCalificación: 5 de 5 estrellas5/5 (2)
- El libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)De EverandEl libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)Calificación: 3 de 5 estrellas3/5 (2)
- Neuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaDe EverandNeuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaCalificación: 4 de 5 estrellas4/5 (16)
- Borges y la memoria: De "Funes el memorioso" a la neurona de Jennifer AnistonDe EverandBorges y la memoria: De "Funes el memorioso" a la neurona de Jennifer AnistonCalificación: 5 de 5 estrellas5/5 (4)
- Cómo hacer aviones de papel y otros objetos voladoresDe EverandCómo hacer aviones de papel y otros objetos voladoresAún no hay calificaciones
- Dieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasDe EverandDieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasCalificación: 4 de 5 estrellas4/5 (9)
- Notas de clase. Manual de farmacognosia: Análisis microscópico y fitoquímico, y usos de plantas medicinalesDe EverandNotas de clase. Manual de farmacognosia: Análisis microscópico y fitoquímico, y usos de plantas medicinalesAún no hay calificaciones
- Zensorialmente : Dejá que tu cuerpo sea tu cerebroDe EverandZensorialmente : Dejá que tu cuerpo sea tu cerebroAún no hay calificaciones
- Trucos para Leer la Mente de los Demás: Cómo Adivinar el Pensamiento de los Demás con Poco Esfuerzo. 2 Libros en 1 - Secretos de la Psicología Oscura, Cómo ser un Detector de MentirasDe EverandTrucos para Leer la Mente de los Demás: Cómo Adivinar el Pensamiento de los Demás con Poco Esfuerzo. 2 Libros en 1 - Secretos de la Psicología Oscura, Cómo ser un Detector de MentirasCalificación: 4.5 de 5 estrellas4.5/5 (3)
- El péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaDe EverandEl péndulo de sanación: Péndulo hebreo. Investigación y sistematización de la técnicaCalificación: 4.5 de 5 estrellas4.5/5 (27)
- El cerebro del niño explicado a los padresDe EverandEl cerebro del niño explicado a los padresCalificación: 4.5 de 5 estrellas4.5/5 (147)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- La invasión de la pseudociencia: Claves para orientarse en un mundo en donde casi todo es posibleDe EverandLa invasión de la pseudociencia: Claves para orientarse en un mundo en donde casi todo es posibleCalificación: 4.5 de 5 estrellas4.5/5 (15)