También podría gustarte
- Ejercicios de Integrales y Ecuaciones Integrales-DiferencialesDe EverandEjercicios de Integrales y Ecuaciones Integrales-DiferencialesAún no hay calificaciones
- Capitulo II - Estática de Los FluidosDocumento49 páginasCapitulo II - Estática de Los FluidosALEJANDRO ALBINO TORPOCO SOTOAún no hay calificaciones
- Español 3 Grado SecundariaDocumento26 páginasEspañol 3 Grado SecundariaDavid Karaokanta ShaggysAún no hay calificaciones
- Cálculo de Gas LibreDocumento38 páginasCálculo de Gas LibreCarlitos BaezAún no hay calificaciones
- ST - apunTES 2. Calderas y Combusti NDocumento44 páginasST - apunTES 2. Calderas y Combusti NHugo Sepulveda MuñozAún no hay calificaciones
- CONCLUSIONES SecadoDocumento1 páginaCONCLUSIONES SecadoJose Luis Barradas100% (1)
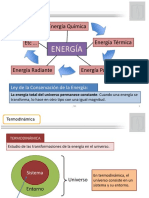
- Sistema Cerrado-TermodinamicaDocumento11 páginasSistema Cerrado-TermodinamicaORIANA ALEXA GUTIERREZ ROJASAún no hay calificaciones
- Problemas Del PathriaDocumento13 páginasProblemas Del PathriaMontse CrespoAún no hay calificaciones
- Equipos SuperficialesDocumento29 páginasEquipos SuperficialesOscar Mamani LopezAún no hay calificaciones
- Termodinámica Aplicada - Trabajo 2020-2Documento46 páginasTermodinámica Aplicada - Trabajo 2020-2Jhonatan Osorio durandAún no hay calificaciones
- Clase2 PDFDocumento7 páginasClase2 PDFMATIAS GABRIEL RICCIAún no hay calificaciones
- Clase 20-5, 2Documento9 páginasClase 20-5, 2Valeria RequenaAún no hay calificaciones
- KARINADocumento10 páginasKARINAhaquilesAún no hay calificaciones
- Flujo Irrotacional - BorradorDocumento11 páginasFlujo Irrotacional - BorradorAinara Cecilia SalaverriaAún no hay calificaciones
- CAPÍTULO TRABAJO Y COLOR - TermodinamicaDocumento11 páginasCAPÍTULO TRABAJO Y COLOR - Termodinamicaapuatau94620% (2)
- Diapositivas Dinamica de Los Fluidos PerfectosDocumento34 páginasDiapositivas Dinamica de Los Fluidos PerfectosYessica Diaz AlbaAún no hay calificaciones
- Trabajo de Expansión de FisicoquimicaDocumento16 páginasTrabajo de Expansión de FisicoquimicaKarlos Pizarro100% (1)
- Practica 4 TermodinamicaDocumento11 páginasPractica 4 TermodinamicaAbner MartinezAún no hay calificaciones
- Primera Ley de TermodinamicaDocumento71 páginasPrimera Ley de TermodinamicaAndreaAún no hay calificaciones
- F.Q 3Documento23 páginasF.Q 3Gabriel AndrésAún no hay calificaciones
- Cap 4. Primera Ley de TermodinámicaDocumento23 páginasCap 4. Primera Ley de TermodinámicaJulieth guzmánAún no hay calificaciones
- Cap 2A 2023Documento20 páginasCap 2A 2023Diego Rodrigo Uribe RodriguezAún no hay calificaciones
- Trabajo TermodinámicoDocumento5 páginasTrabajo Termodinámicobyron guerreroAún no hay calificaciones
- 1ra Ley Proc Fisicos - Sin Audios - CDocumento56 páginas1ra Ley Proc Fisicos - Sin Audios - CGC JhoselinAún no hay calificaciones
- Semana N°3 - Primera Ley de La TermodinámicaDocumento20 páginasSemana N°3 - Primera Ley de La TermodinámicaLesly PeredaAún no hay calificaciones
- Ley y Ecuacion EnergeticaDocumento19 páginasLey y Ecuacion EnergeticaMayra VeraAún no hay calificaciones
- Sesión - 04 - Calor y Trabajo en TermodinámicaDocumento17 páginasSesión - 04 - Calor y Trabajo en TermodinámicaRenato Carlos PulidoAún no hay calificaciones
- Trabajo y Calor NuevaDocumento40 páginasTrabajo y Calor NuevaMayra Leal75% (4)
- Aplicacion de ED de Orden SuperionDocumento28 páginasAplicacion de ED de Orden SuperionStalyn PilamungaAún no hay calificaciones
- Calor y TrabajoDocumento53 páginasCalor y TrabajoCarlos Hugo Graverolle LópezAún no hay calificaciones
- Primera Ley Problemas Resueltos TermodinamicaDocumento27 páginasPrimera Ley Problemas Resueltos TermodinamicaJuvenal GalarceAún no hay calificaciones
- 3-Primera LeyDocumento52 páginas3-Primera LeySofi RomeroAún no hay calificaciones
- PrimeraleydelatermodinmicaDocumento65 páginasPrimeraleydelatermodinmicaKaren CanteroAún no hay calificaciones
- Mecánica Clásica: Breviario de la Mecánica Clásica siguiendo a H. GoldsteinDocumento114 páginasMecánica Clásica: Breviario de la Mecánica Clásica siguiendo a H. GoldsteinAgustina ZapataAún no hay calificaciones
- Semana 1ADocumento14 páginasSemana 1AElor TorrotoAún no hay calificaciones
- Clase 3.1 Trabajo Termodinámico P-VDocumento7 páginasClase 3.1 Trabajo Termodinámico P-VVale IturraAún no hay calificaciones
- Resumen de Mecánica de Los FluidosDocumento80 páginasResumen de Mecánica de Los FluidosJulian SarmientoAún no hay calificaciones
- Primer Principio de La TermodinámicaDocumento27 páginasPrimer Principio de La TermodinámicaJavier Hans Zapata EspinozaAún no hay calificaciones
- Primera Ley de La Termodinamica AplicadaDocumento4 páginasPrimera Ley de La Termodinamica AplicadaJoseph Condori CahuiAún no hay calificaciones
- Flujo SanguineoDocumento65 páginasFlujo Sanguineoapi-371168786% (7)
- Formato APAGeneralDocumento19 páginasFormato APAGeneralAlexis Marca MaqueraAún no hay calificaciones
- Fisicoquimica IDocumento224 páginasFisicoquimica IMANUEL SANCHEZ100% (1)
- EXPANSION Y COMPRESION TeoricoDocumento9 páginasEXPANSION Y COMPRESION TeoricoflamarrionAún no hay calificaciones
- Trabajo Virtual Como La Forma Debil de Las Ecuaciones de Equilibrio para El Analisis de Solidos O FluidosDocumento9 páginasTrabajo Virtual Como La Forma Debil de Las Ecuaciones de Equilibrio para El Analisis de Solidos O FluidosHéctor FuentesAún no hay calificaciones
- Apuntes Principios de MáquinasDocumento37 páginasApuntes Principios de MáquinasaratecnoAún no hay calificaciones
- Principio de los Trabajos Virtuales en EstructurasDocumento8 páginasPrincipio de los Trabajos Virtuales en Estructurasalbertito17Aún no hay calificaciones
- Semana 10. TermoquimicaDocumento39 páginasSemana 10. TermoquimicaAlexander DuránAún no hay calificaciones
- FISICA I U VI Trabajo y EnergiaDocumento13 páginasFISICA I U VI Trabajo y EnergiaLalo MorenoAún no hay calificaciones
- Diapositivas Dinamica de Los Fluidos Perfectos ArregladasDocumento36 páginasDiapositivas Dinamica de Los Fluidos Perfectos ArregladasRicardo Requejo CarrilloAún no hay calificaciones
- TF-1121 Guía de Estudio Teoria y Problemas Capitulo 3Documento33 páginasTF-1121 Guía de Estudio Teoria y Problemas Capitulo 3Oriana Gudiño GaliñoAún no hay calificaciones
- HidrostaticaDocumento17 páginasHidrostaticaIndi Huamanvilca HuamaniAún no hay calificaciones
- Capitulo II - Mecanica de FluidosDocumento14 páginasCapitulo II - Mecanica de FluidosJOHN FELIX ESQUIVEL RAMOSAún no hay calificaciones
- Energía de Los Fluidos IncompresiblesDocumento7 páginasEnergía de Los Fluidos IncompresiblesRauloxzAún no hay calificaciones
- Respuestas Experimento Gato Hidraulico Pedro Goris 2019 7633 PDFDocumento4 páginasRespuestas Experimento Gato Hidraulico Pedro Goris 2019 7633 PDFHector GuerraAún no hay calificaciones
- TERMO1-TEMA5 Trabajo y CalorDocumento30 páginasTERMO1-TEMA5 Trabajo y CalorLuis Alejandro GuillermoAún no hay calificaciones
- Resumen de TermoquímicaDocumento20 páginasResumen de TermoquímicaValentino FiccaAún no hay calificaciones
- Estática de fluidos en reposoDocumento29 páginasEstática de fluidos en reposoEvelin Karla Mamani ArroyoAún no hay calificaciones
- Capitulo II - Estática de Los FluidosDocumento38 páginasCapitulo II - Estática de Los FluidosEvelin Karla Mamani ArroyoAún no hay calificaciones
- TermoDocumento15 páginasTermoJean carlos Patala sanchezAún no hay calificaciones
- Aprende Que Es La Tensión SuperficialDocumento2 páginasAprende Que Es La Tensión SuperficialJulio César DoloresAún no hay calificaciones
- Identidad Cultural en MéxicoDocumento19 páginasIdentidad Cultural en MéxicoJulio César DoloresAún no hay calificaciones
- Ejercicio Función BUSCARVDocumento5 páginasEjercicio Función BUSCARVJulio César DoloresAún no hay calificaciones
- Control calidad obra 3 pasosDocumento1 páginaControl calidad obra 3 pasosJulio César DoloresAún no hay calificaciones
- Unidad 3Documento22 páginasUnidad 3Julio César DoloresAún no hay calificaciones
- Equipo 2.1Documento5 páginasEquipo 2.1Julio César DoloresAún no hay calificaciones
- Fotos de Quimica GeneralDocumento12 páginasFotos de Quimica GeneralJulio César DoloresAún no hay calificaciones
- BVP4C MatlabDocumento11 páginasBVP4C MatlabbarbarojAún no hay calificaciones
- Archivo 20201228122433Documento10 páginasArchivo 20201228122433WILLIAM FRANCISCO MENDIETA BENAVIDESAún no hay calificaciones
- Estado GaseosoDocumento27 páginasEstado Gaseosoomar kanaAún no hay calificaciones
- CAP ìTULODocumento50 páginasCAP ìTULOKEVINAún no hay calificaciones
- Estado LiquidoDocumento24 páginasEstado Liquidoc2fng8rcmtAún no hay calificaciones
- EstadosMateriaSEODocumento4 páginasEstadosMateriaSEOChRis DiiaZzAún no hay calificaciones
- Yacimiento Sub SaturadoDocumento11 páginasYacimiento Sub SaturadoElvis HuarayoAún no hay calificaciones
- Tecnologias de Deshidratacion Del Gas AnturalDocumento7 páginasTecnologias de Deshidratacion Del Gas Anturalsanty222Aún no hay calificaciones
- 3eso U02Documento14 páginas3eso U02emmanuelAún no hay calificaciones
- Guia de Matematicas Grado Tercero, Tercer PeriodoDocumento15 páginasGuia de Matematicas Grado Tercero, Tercer PeriodoEnnailen LaitonAún no hay calificaciones
- PRACTICA 2 - 4to ParcialDocumento5 páginasPRACTICA 2 - 4to ParcialRodriguez MamaniAún no hay calificaciones
- CAPITULO 4 Tecnicas para El Control de ArenaDocumento11 páginasCAPITULO 4 Tecnicas para El Control de ArenaJavier RiosAún no hay calificaciones
- Ejemplos Cotidianos de Las Caracteristicas de Los GasesDocumento1 páginaEjemplos Cotidianos de Las Caracteristicas de Los GasesLeonardo Daniel Lugo GastelumAún no hay calificaciones
- Taller. Estequiometria de GasesDocumento8 páginasTaller. Estequiometria de GasesJENI MARCILLO MORÁNAún no hay calificaciones
- Trabajo de Investigacion, Hidraulica. Angeles RamirezDocumento14 páginasTrabajo de Investigacion, Hidraulica. Angeles RamirezLuis Alfonso Ramirez LozanoAún no hay calificaciones
- HDS NitrogenoDocumento3 páginasHDS Nitrogenofede hoyosAún no hay calificaciones
- Diseño de Biodigestor en CubaDocumento28 páginasDiseño de Biodigestor en CubaAnonymous j2YLxJVi5cAún no hay calificaciones
- 3°QuímicaBloque2Secuencia11 12sesiones11,1 5Documento6 páginas3°QuímicaBloque2Secuencia11 12sesiones11,1 5Julieta GutlAún no hay calificaciones
- Ciclo HexanoDocumento131 páginasCiclo HexanoMeliza Rosi Bravo CastilloAún no hay calificaciones
- INFORME Ley de BoyleDocumento4 páginasINFORME Ley de BoyleDiego A. Casas Aquino67% (3)
- Silabo MF1 Usat 2013-1Documento4 páginasSilabo MF1 Usat 2013-1Kevyn Đávila BurgaAún no hay calificaciones
- Universidad Nacional de Ancash Santiago Antúnez de MayoloDocumento14 páginasUniversidad Nacional de Ancash Santiago Antúnez de MayoloKiara Jursiny Villenueva DominguezAún no hay calificaciones
- Apuntes de Productividad de PozosDocumento34 páginasApuntes de Productividad de PozosAndrésTorresAún no hay calificaciones
- Película de Alta Barrera de 7 Capas para Empaque de AlimentosDocumento1 páginaPelícula de Alta Barrera de 7 Capas para Empaque de Alimentoseded_ls_caAún no hay calificaciones
- Ejercicios de mecánica de fluidos y sus propiedadesDocumento2 páginasEjercicios de mecánica de fluidos y sus propiedadesDilmer VelasquezAún no hay calificaciones