También podría gustarte
- Introducción a la Biología: RESÚMENES UNIVERSITARIOSDe EverandIntroducción a la Biología: RESÚMENES UNIVERSITARIOSCalificación: 5 de 5 estrellas5/5 (1)
- La PCR Trabajo PDFDocumento16 páginasLa PCR Trabajo PDFOmar MohamedAún no hay calificaciones
- Tecnicas o Herramientas para El Estudio de DnaDocumento18 páginasTecnicas o Herramientas para El Estudio de Dnaanon_355220885Aún no hay calificaciones
- PCRDocumento6 páginasPCREs BobyyAún no hay calificaciones
- Reacción en Cadena de La PolimerasaDocumento9 páginasReacción en Cadena de La PolimerasasilviaAún no hay calificaciones
- Practica 2. ReporteDocumento11 páginasPractica 2. ReportePorta MuñozAún no hay calificaciones
- 41 Comprobación Colonias Transformantes PCRDocumento5 páginas41 Comprobación Colonias Transformantes PCRLednys Palomino ZambranoAún no hay calificaciones
- Reaccion en Cadena de La Polimerasa CelularDocumento1 páginaReaccion en Cadena de La Polimerasa Celularangela morgadoAún no hay calificaciones
- PCR Lab GenDocumento7 páginasPCR Lab GenPatricio pincheiraAún no hay calificaciones
- PCR ConvencionalDocumento3 páginasPCR Convencionalcassievw314Aún no hay calificaciones
- TALLER DE REACCIÓN EN CADENA DE LA POLIMERASA (PCR) (1) (2)Documento5 páginasTALLER DE REACCIÓN EN CADENA DE LA POLIMERASA (PCR) (1) (2)Rosa Isabel Guette TorneAún no hay calificaciones
- Control 5 Biocel Lab ?Documento7 páginasControl 5 Biocel Lab ?Florencia Antonia Silva HenríquezAún no hay calificaciones
- Informe G&e-S10Documento8 páginasInforme G&e-S10MARIA FERNANDA PINEDO AZA�EROAún no hay calificaciones
- ¿Qué Es La PCR?: Secuenciar Electroforesis en Gel ClonarDocumento3 páginas¿Qué Es La PCR?: Secuenciar Electroforesis en Gel Clonarvaleriasantoyo289Aún no hay calificaciones
- Ayuda MemoriaDocumento4 páginasAyuda MemoriaKarina QuintoAún no hay calificaciones
- Diapo 1Documento4 páginasDiapo 1JUANAún no hay calificaciones
- Caso Tecnicas MolecularesDocumento4 páginasCaso Tecnicas MolecularesMilena RodriguezAún no hay calificaciones
- 41 Comprobación Colonias Transformantes PCRDocumento5 páginas41 Comprobación Colonias Transformantes PCRlevymiguel3012726Aún no hay calificaciones
- RFLP y PCRDocumento2 páginasRFLP y PCRAlexander Casian PlazaAún no hay calificaciones
- Tecnicas Moleculares para Replicación DNADocumento7 páginasTecnicas Moleculares para Replicación DNAMelissa GalanAún no hay calificaciones
- 5 PCRDocumento6 páginas5 PCRJose EnriqueAún no hay calificaciones
- Taller Biotecnología UnalDocumento4 páginasTaller Biotecnología UnalRodolfo AriasAún no hay calificaciones
- Herramientas Moleculares 27Documento14 páginasHerramientas Moleculares 27mana taraAún no hay calificaciones
- Lab MolecularDocumento4 páginasLab MolecularYessica OteroAún no hay calificaciones
- Amplificación Del ADN-Salazar LucioDocumento6 páginasAmplificación Del ADN-Salazar LucioNoel Îa SalazarAún no hay calificaciones
- PCR Reacción en Cadena de La PolimerasaDocumento10 páginasPCR Reacción en Cadena de La PolimerasaJessica Hernandez SanchezAún no hay calificaciones
- REPLICACIÓN Y PCR (Control Lab, Materia Solemne)Documento12 páginasREPLICACIÓN Y PCR (Control Lab, Materia Solemne)Yisenia VillegasAún no hay calificaciones
- Aplicaciones de La PCRDocumento4 páginasAplicaciones de La PCRYanine HikariAún no hay calificaciones
- PCR Anidada: Técnica Muy Sensible de PCR en La Que El Producto de Una Amplificación EsDocumento12 páginasPCR Anidada: Técnica Muy Sensible de PCR en La Que El Producto de Una Amplificación EsValeria BoomAún no hay calificaciones
- Seminario 6 BiologíaDocumento18 páginasSeminario 6 BiologíaDafne Olenka Davila PerezAún no hay calificaciones
- PCR - Reaccion en Cadena de La Polimerasa y AluDocumento9 páginasPCR - Reaccion en Cadena de La Polimerasa y AluArantxa González IturraAún no hay calificaciones
- Informe Gye Semana 10Documento8 páginasInforme Gye Semana 10KIARA ANALY SHOCUSH ARMASAún no hay calificaciones
- Qué Es La PCRDocumento4 páginasQué Es La PCRCamila ParraAún no hay calificaciones
- BioMol TareaDocumento8 páginasBioMol TareaMARIA FERNANDA VICENTTIN PERUSQUIAAún no hay calificaciones
- GE Reaccion en Cadena de La Polimerasa PCRDocumento11 páginasGE Reaccion en Cadena de La Polimerasa PCRFlorencia MerinoAún no hay calificaciones
- ADN ForenseDocumento5 páginasADN ForenseGonzalo AguilarAún no hay calificaciones
- Tecnicas y Marcadores de Biologia MolecularDocumento9 páginasTecnicas y Marcadores de Biologia MolecularCesar GutierrezAún no hay calificaciones
- La Hibridación de Ácidos NucleicosDocumento11 páginasLa Hibridación de Ácidos NucleicosLaura KatherinAún no hay calificaciones
- Reacciã"n en Cadena de La Polimerasa FinalDocumento33 páginasReacciã"n en Cadena de La Polimerasa FinalJOSUEAún no hay calificaciones
- Identificación de Ácidos NucleicosDocumento6 páginasIdentificación de Ácidos NucleicosTania GonzalezAún no hay calificaciones
- Caso 2 BCMDocumento5 páginasCaso 2 BCMCecilia CaiafaAún no hay calificaciones
- Dna Polimórfico Amplificado Al AzarDocumento4 páginasDna Polimórfico Amplificado Al AzarJosé PaterninaAún no hay calificaciones
- Documento Sin TítuloDocumento4 páginasDocumento Sin TítuloRuben GuardiaAún no hay calificaciones
- PCR-RT: Amplificación de ARNDocumento9 páginasPCR-RT: Amplificación de ARNvioletaAún no hay calificaciones
- 4-Estudio de Las Mutaciones Relacionadas Con El Cáncer para Establecer La Terapia Adecuada-2Documento1 página4-Estudio de Las Mutaciones Relacionadas Con El Cáncer para Establecer La Terapia Adecuada-2Marieta GarcíaAún no hay calificaciones
- Informe FinalDocumento17 páginasInforme FinalRenzo AvilaAún no hay calificaciones
- Kit de Huella Genetica (ADN Fingerprint) PDFDocumento41 páginasKit de Huella Genetica (ADN Fingerprint) PDFAndres Duran100% (1)
- Cuestiones de Aplicación PCRDocumento8 páginasCuestiones de Aplicación PCRLara Vázquez FafiánAún no hay calificaciones
- Reaccion de Cadena de PolimerasaDocumento27 páginasReaccion de Cadena de PolimerasaAndrea SolanoAún no hay calificaciones
- Informe PCRDocumento5 páginasInforme PCRxxxAún no hay calificaciones
- Exposición de MolecularDocumento37 páginasExposición de Molecularmelissa cuellarAún no hay calificaciones
- PCR-UG-2022Documento14 páginasPCR-UG-2022Vanessa AguiñoAún no hay calificaciones
- Informe Exposición Grupo N2 BioinformáticaDocumento4 páginasInforme Exposición Grupo N2 BioinformáticaNADIA NICOLE CELI BOHORQUEZAún no hay calificaciones
- Laboratorio Pcr-BiologíaDocumento11 páginasLaboratorio Pcr-BiologíaPaula Andrea MoraAún no hay calificaciones
- PCR InformeDocumento6 páginasPCR InformeSergio PinedoAún no hay calificaciones
- Trabajo Final PCR y Secuenciación - TiradoZazuetaGuadalupeDocumento10 páginasTrabajo Final PCR y Secuenciación - TiradoZazuetaGuadalupeGuadalupe TZAún no hay calificaciones
- Cours PDFDocumento91 páginasCours PDFMencia Fuente CastrilloAún no hay calificaciones
- Herramienta Moleculares y Su Aplicación ClínicaDocumento15 páginasHerramienta Moleculares y Su Aplicación ClínicaZUHEIRY PCAún no hay calificaciones
- ElectroforesisDocumento17 páginasElectroforesisAdrian EspejelAún no hay calificaciones
- Monografia Biosintesis de ProteinasDocumento23 páginasMonografia Biosintesis de ProteinasSoberon BernabeAún no hay calificaciones
- Cuestionario Capitulo 11Documento7 páginasCuestionario Capitulo 11Elizabeth LC100% (1)
- Examen de Biología Grado 9ºDocumento1 páginaExamen de Biología Grado 9ºnorismercadoAún no hay calificaciones
- Preguntas y Respuestas PolimerosDocumento64 páginasPreguntas y Respuestas PolimerosVale Mendez100% (2)
- Estructuras de Las Proteinas - ADocumento25 páginasEstructuras de Las Proteinas - AClarisa CarriónAún no hay calificaciones
- Retículo Endoplásmicogolgi-2Documento60 páginasRetículo Endoplásmicogolgi-2Bastián HurtadoAún no hay calificaciones
- CLASE 15. Ácidos Nucleicos COMPLETADocumento45 páginasCLASE 15. Ácidos Nucleicos COMPLETASaul GonzalezAún no hay calificaciones
- Taller Acidosnucleicos UCMDocumento3 páginasTaller Acidosnucleicos UCMBerrio Martinez LocoAún no hay calificaciones
- Marcadores EnzimaticosDocumento4 páginasMarcadores EnzimaticosFiorella Lizzeth GpAún no hay calificaciones
- 11taller de GeneticaDocumento4 páginas11taller de GeneticaMARLON ALEXANDER CONTRERAS LOPEZ100% (1)
- Guia Biologia - Iv° MedioDocumento6 páginasGuia Biologia - Iv° MedioRoberto PachecoAún no hay calificaciones
- ListadesustanciasderellenoprohibidasDocumento12 páginasListadesustanciasderellenoprohibidasPatricia Mercedes Hernández FloresAún no hay calificaciones
- Informe de LaboratorioDocumento10 páginasInforme de LaboratorioNicole Carolina Quintero RomeroAún no hay calificaciones
- 2017 02 04 12 PDFDocumento4 páginas2017 02 04 12 PDFAman MinjAún no hay calificaciones
- TranscripcionDocumento8 páginasTranscripcionDeboraAún no hay calificaciones
- Colesterol HDL CalibradorDocumento1 páginaColesterol HDL CalibradorRoger Quispe RiquelmeAún no hay calificaciones
- Mapa NucleoDocumento1 páginaMapa NucleoLuis Eduardo de Paz BarreraAún no hay calificaciones
- Exposicion 3Documento9 páginasExposicion 3bruce vegaAún no hay calificaciones
- Practica Virtual Montaje de La Estructura Del Adn en Papel y Verificacion de La Secuencia Genetica en El Blastn - Alina Velasque GutierrezDocumento9 páginasPractica Virtual Montaje de La Estructura Del Adn en Papel y Verificacion de La Secuencia Genetica en El Blastn - Alina Velasque GutierrezAlina Velasque GutierrezAún no hay calificaciones
- Biologia Tarea 4Documento9 páginasBiologia Tarea 4Michell Ramirez100% (1)
- Cuestionario 4. Enzimas.Documento10 páginasCuestionario 4. Enzimas.andrea rochaAún no hay calificaciones
- Qué Son ADN y ARNDocumento14 páginasQué Son ADN y ARNAdrihel Paolo Ku Aguilar100% (1)
- Cuadro ComparativoDocumento1 páginaCuadro ComparativoMiiguel RodriguezAún no hay calificaciones
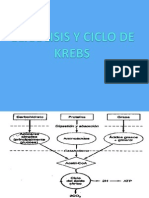
- Glicolisis y Ciclo de KrebsDocumento31 páginasGlicolisis y Ciclo de Krebsfmh2009100% (1)
- Guia de Estudio CitoesqueletoDocumento9 páginasGuia de Estudio CitoesqueletoCarlosAgostoAún no hay calificaciones
- UA4 Aislamiento y Clonado de Genes - AlumnadoDocumento18 páginasUA4 Aislamiento y Clonado de Genes - AlumnadoLe BlauAún no hay calificaciones
- PCR FundamentosDocumento23 páginasPCR FundamentosINGRID MARGARITA GUERRERO MARTINEZAún no hay calificaciones
- Natualeza de Las Moleculas Biologicas PDFDocumento12 páginasNatualeza de Las Moleculas Biologicas PDFCale MillanAún no hay calificaciones
- TRANSCRIPCIONDocumento47 páginasTRANSCRIPCIONAlexa GarroniAún no hay calificaciones
- Semana 14 - PRÁCTICA - Extracción Del ADNDocumento18 páginasSemana 14 - PRÁCTICA - Extracción Del ADNrodrigo blanco barrantesAún no hay calificaciones
- Resumen de Pensar rápido pensar despacio de Daniel KahnemanDe EverandResumen de Pensar rápido pensar despacio de Daniel KahnemanCalificación: 4.5 de 5 estrellas4.5/5 (11)
- En busca de la mente: El largo camino para comprender la vida mental (y lo que aún queda por descubrir)De EverandEn busca de la mente: El largo camino para comprender la vida mental (y lo que aún queda por descubrir)Calificación: 4.5 de 5 estrellas4.5/5 (3)
- Batidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoDe EverandBatidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoCalificación: 5 de 5 estrellas5/5 (2)
- Genética general: Libro de textoDe EverandGenética general: Libro de textoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Abrázame fuerte: Siete conversaciones para un amor duraderoDe EverandAbrázame fuerte: Siete conversaciones para un amor duraderoManu BerásteguiCalificación: 4.5 de 5 estrellas4.5/5 (13)
- 200 tareas en terapia breve: 2ª ediciónDe Everand200 tareas en terapia breve: 2ª ediciónCalificación: 4.5 de 5 estrellas4.5/5 (33)
- 50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaDe Everand50 técnicas de mindfulness para la ansiedad, la depresión, el estrés y el dolor: Mindfulness como terapiaCalificación: 4 de 5 estrellas4/5 (37)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- El autismo: Reflexiones y pautas para comprenderlo y abordarloDe EverandEl autismo: Reflexiones y pautas para comprenderlo y abordarloCalificación: 4 de 5 estrellas4/5 (7)
- Sesgos Cognitivos: Una Fascinante Mirada dentro de la Psicología Humana y los Métodos para Evitar la Disonancia Cognitiva, Mejorar sus Habilidades para Resolver Problemas y Tomar Mejores DecisionesDe EverandSesgos Cognitivos: Una Fascinante Mirada dentro de la Psicología Humana y los Métodos para Evitar la Disonancia Cognitiva, Mejorar sus Habilidades para Resolver Problemas y Tomar Mejores DecisionesCalificación: 4.5 de 5 estrellas4.5/5 (13)
- Terapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaDe EverandTerapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaCalificación: 4.5 de 5 estrellas4.5/5 (11)
- El cerebro del niño explicado a los padresDe EverandEl cerebro del niño explicado a los padresCalificación: 4.5 de 5 estrellas4.5/5 (147)
- Manual técnico de refrigerantesDe EverandManual técnico de refrigerantesCalificación: 4 de 5 estrellas4/5 (4)
- Neuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaDe EverandNeuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaCalificación: 4 de 5 estrellas4/5 (16)
- Plan De Dieta Para La Resistencia A La Insulina & Sistema Inmunológico En EspañolDe EverandPlan De Dieta Para La Resistencia A La Insulina & Sistema Inmunológico En EspañolCalificación: 3.5 de 5 estrellas3.5/5 (2)
- Despeja Tu Mente: Como Dejar De Pensar Demasiado, Vencer A Tu Crítico Interno, Y Replantear Tus Pensamientos Negativos Con Hábitos SaludablesDe EverandDespeja Tu Mente: Como Dejar De Pensar Demasiado, Vencer A Tu Crítico Interno, Y Replantear Tus Pensamientos Negativos Con Hábitos SaludablesAún no hay calificaciones
- Diccionario de neurociencias: Aplicadas a organizaciones y personasDe EverandDiccionario de neurociencias: Aplicadas a organizaciones y personasCalificación: 5 de 5 estrellas5/5 (4)
- Anatomía del caballo: Guía práctica ilustradaDe EverandAnatomía del caballo: Guía práctica ilustradaCalificación: 4 de 5 estrellas4/5 (9)
- Mujeres Con Trastornos Por Déficit De Atención: Cómo aceptar sus diferencias y transformar su vidaDe EverandMujeres Con Trastornos Por Déficit De Atención: Cómo aceptar sus diferencias y transformar su vidaCalificación: 3.5 de 5 estrellas3.5/5 (3)
- Bases biológicas del comportamiento animal y humanoDe EverandBases biológicas del comportamiento animal y humanoCalificación: 4 de 5 estrellas4/5 (4)
- Principios básicos de bioquímica de los alimentosDe EverandPrincipios básicos de bioquímica de los alimentosCalificación: 4.5 de 5 estrellas4.5/5 (2)
- Medicina con plantas sagradas: La sabiduría del herbalismo de los aborígenes norteamericanosDe EverandMedicina con plantas sagradas: La sabiduría del herbalismo de los aborígenes norteamericanosCalificación: 4 de 5 estrellas4/5 (10)