También podría gustarte
- Sindrome de DownDocumento56 páginasSindrome de DownoptoipncicsstAún no hay calificaciones
- MESA RADIÔNICA SISTÊMICA - Ebook - 2022 NOVIEMBRE TEORICODocumento121 páginasMESA RADIÔNICA SISTÊMICA - Ebook - 2022 NOVIEMBRE TEORICOConita-Boyd75% (4)
- Pubertad PrecozDocumento16 páginasPubertad PrecozRuth BurgaAún no hay calificaciones
- Manual de Estrategias para Personas Con Paralisis CerebralDocumento21 páginasManual de Estrategias para Personas Con Paralisis CerebralJym Carlos100% (1)
- Diapo Pediatria 1-FinalDocumento49 páginasDiapo Pediatria 1-FinalAndru Salazar100% (1)
- Modelo Centrado en La TareaDocumento18 páginasModelo Centrado en La Tareajanndery soncco basauriAún no hay calificaciones
- Taller SX de DownDocumento8 páginasTaller SX de DownYubelly Andrea Rodriguez ChavrroAún no hay calificaciones
- Síndromes GenéticosDocumento6 páginasSíndromes GenéticosDavid Hernández SotoAún no hay calificaciones
- Síndromes Cromosómicas - PRÁCTICA N°02 E.GDocumento19 páginasSíndromes Cromosómicas - PRÁCTICA N°02 E.GClaudia Mendoza VarasAún no hay calificaciones
- Presentación 2 Genética y ComportamientoDocumento16 páginasPresentación 2 Genética y ComportamientoYaneth AguilarAún no hay calificaciones
- 6 Down With Cover Page v2Documento8 páginas6 Down With Cover Page v2Erick MenesesAún no hay calificaciones
- Alteraciones Cromosómicas AutosómicasDocumento7 páginasAlteraciones Cromosómicas AutosómicasMaria PerezAún no hay calificaciones
- Síndrome de DownDocumento152 páginasSíndrome de DownChristianCamiloMartínezAún no hay calificaciones
- Seminario de Neurodesarrollo y Sus DesórdenesDocumento23 páginasSeminario de Neurodesarrollo y Sus DesórdenesMatias DurandAún no hay calificaciones
- Caso Genetica Semana 3Documento6 páginasCaso Genetica Semana 3andreaAún no hay calificaciones
- Down, Turner, FibrosisDocumento25 páginasDown, Turner, FibrosisAnónimoAún no hay calificaciones
- Síndrome de Down.Documento16 páginasSíndrome de Down.LIZBETH ANNER SANTIAGO ECHEVARRIAAún no hay calificaciones
- Sindrome de DownDocumento5 páginasSindrome de DownJhenny ZarateAún no hay calificaciones
- Alteraciones CromosomicasDocumento7 páginasAlteraciones CromosomicasAndres UlloaAún no hay calificaciones
- Sindromes Geneticos Expo Pediatria Mip AlmeidaDocumento71 páginasSindromes Geneticos Expo Pediatria Mip AlmeidaNicolas Andres Rivera GuenchurAún no hay calificaciones
- Cariotipo 3 CorteDocumento10 páginasCariotipo 3 CorteJudith Miranda PoloAún no hay calificaciones
- CromosomopatiasDocumento15 páginasCromosomopatiasDavid Guillermo Gallo GarcíaAún no hay calificaciones
- Deteccion Selectiva de AneuploidiasDocumento87 páginasDeteccion Selectiva de Aneuploidiasdra.lucypanrxAún no hay calificaciones
- Pediatrai III-clase 5Documento8 páginasPediatrai III-clase 5Liam MartinezAún no hay calificaciones
- Eco GenéticoDocumento33 páginasEco GenéticoClauMcClauAún no hay calificaciones
- Síndrome de PatauDocumento17 páginasSíndrome de PatauYesleni OrozcoAún no hay calificaciones
- Talla Baja y Talla AltaDocumento16 páginasTalla Baja y Talla AltaGerardo100% (1)
- Nº6 Teoría Genética Resuelto Semana 6 UpaoDocumento7 páginasNº6 Teoría Genética Resuelto Semana 6 UpaoMaría Gracia MachadoAún no hay calificaciones
- Mehu525 U5 T14 Tamizaje Neonatal y CromosomopatíasDocumento28 páginasMehu525 U5 T14 Tamizaje Neonatal y CromosomopatíasAndreaAún no hay calificaciones
- Aberraciones CromosómicasDocumento13 páginasAberraciones Cromosómicasfrank150100% (2)
- ChongW - DelgadoJ - GalarragaV - GutierrezM - Pràctica de Aplicaciòn - Síndrome de EdwardsDocumento3 páginasChongW - DelgadoJ - GalarragaV - GutierrezM - Pràctica de Aplicaciòn - Síndrome de EdwardsWinstong ChongAún no hay calificaciones
- Enfermedades Geneticas en Recien NacidosDocumento72 páginasEnfermedades Geneticas en Recien NacidosRuby CaraballoAún no hay calificaciones
- Laboratorio Iii Grupo 4 EmbriologíaDocumento7 páginasLaboratorio Iii Grupo 4 EmbriologíaRos Barboza100% (1)
- CASO CLÍNICO SINDROME DE DOWN-Grupo 2Documento12 páginasCASO CLÍNICO SINDROME DE DOWN-Grupo 2Pao Miranda BaronaAún no hay calificaciones
- 3p25 Deleciones 3p25 Tambien Llamadas 3p - (Menos) Spanish FTNPDocumento6 páginas3p25 Deleciones 3p25 Tambien Llamadas 3p - (Menos) Spanish FTNPYOHANA TORRESAún no hay calificaciones
- Patología General I - Entrega de Producto Ii - HemangiomaDocumento16 páginasPatología General I - Entrega de Producto Ii - HemangiomaKarolTorresHAún no hay calificaciones
- Criptorquidia - Escala de GleasonDocumento27 páginasCriptorquidia - Escala de GleasonCamille KirschbaumAún no hay calificaciones
- Enfermedades Pediátricas.Documento5 páginasEnfermedades Pediátricas.Ariana Francella Mercado LópezAún no hay calificaciones
- Sindrome de DownDocumento24 páginasSindrome de DownSandiAún no hay calificaciones
- Caso Clínico DiplomadoDocumento20 páginasCaso Clínico DiplomadoJose Gregorio Gutiérrez OAún no hay calificaciones
- Caso Clinico Sindrome de Down PDFDocumento15 páginasCaso Clinico Sindrome de Down PDFernesto balladares100% (1)
- SindromedeDown AntecedentesDocumento9 páginasSindromedeDown AntecedentesMarAún no hay calificaciones
- CROMOSOMOPATIADocumento13 páginasCROMOSOMOPATIAFrp CuentasAún no hay calificaciones
- 66-71 Trisomia 8Documento6 páginas66-71 Trisomia 8Giovanni MondragonAún no hay calificaciones
- 6 DownDocumento7 páginas6 DownValentina GarciaAún no hay calificaciones
- SINDROMESDocumento17 páginasSINDROMESJorge Arteaga TorrezAún no hay calificaciones
- Pediatria ForenseDocumento57 páginasPediatria ForenseMarivel Silvia Pulido LeonAún no hay calificaciones
- Sminario de NeurodesarrolloDocumento14 páginasSminario de NeurodesarrolloMatias DurandAún no hay calificaciones
- SX Por AlteracionDocumento65 páginasSX Por AlteracionEdgar Mart Garcia GonzalezAún no hay calificaciones
- Aberraciones Cromosómicas de Número Sindrome de Down - Grupo 3Documento11 páginasAberraciones Cromosómicas de Número Sindrome de Down - Grupo 3estebanmaranon251Aún no hay calificaciones
- Sindrome de Down: Génetica Medica J. Jesús Franco AguirreDocumento21 páginasSindrome de Down: Génetica Medica J. Jesús Franco AguirreJ Jesus Franco AguirreAún no hay calificaciones
- Examen Físico Por Aparatos y Sistemas 2Documento22 páginasExamen Físico Por Aparatos y Sistemas 2Helen Domenica Apolo SigchoAún no hay calificaciones
- Trisomia 13 y 18Documento4 páginasTrisomia 13 y 18Moira Contreras RojasAún no hay calificaciones
- Neonato PrematuroDocumento39 páginasNeonato Prematuroclaubor100% (1)
- DMFSH CT 4Documento55 páginasDMFSH CT 4anamarAún no hay calificaciones
- Trastornos de Los AutosomasDocumento73 páginasTrastornos de Los AutosomasAlluka SacAún no hay calificaciones
- Niño DismórficoDocumento77 páginasNiño Dismórficojdelta28Aún no hay calificaciones
- Sindrome DownDocumento7 páginasSindrome DownDennis Quispe PuchocAún no hay calificaciones
- Alteraciones CromosomicasDocumento42 páginasAlteraciones Cromosomicasalaindr849Aún no hay calificaciones
- Clase 1-7Documento251 páginasClase 1-7Alicia AliagaAún no hay calificaciones
- Acne en El Recien NacidoDocumento7 páginasAcne en El Recien NacidoAntony VacaAún no hay calificaciones
- Síndrome de DownDocumento5 páginasSíndrome de DownmaininimariaeugeniaAún no hay calificaciones
- Didáctica, Organización Escolar y Educación EspecialDocumento72 páginasDidáctica, Organización Escolar y Educación EspecialBertha Martinez ZamoraAún no hay calificaciones
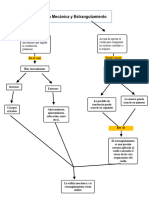
- Mapa Conceptual Asfixia Mecanica y EstrangulamientoDocumento2 páginasMapa Conceptual Asfixia Mecanica y Estrangulamientogalindogenesis26Aún no hay calificaciones
- Reconociendo Los Escenarios de La Educación Superior Mexicana Durante La Pandemia.Documento180 páginasReconociendo Los Escenarios de La Educación Superior Mexicana Durante La Pandemia.QartuppiAún no hay calificaciones
- Mapa Conceptual Dermatitis AtópicaDocumento1 páginaMapa Conceptual Dermatitis Atópicakaren50% (2)
- Vida LaboralDocumento4 páginasVida LaboralMaria CamarenaAún no hay calificaciones
- Fisiologia Del PartoDocumento73 páginasFisiologia Del PartoPiere AcharteAún no hay calificaciones
- Npnu U2 Atr AllaDocumento3 páginasNpnu U2 Atr AllaAlfonso Lopez AvilaAún no hay calificaciones
- RisperidonaDocumento116 páginasRisperidonaAguirre Milka100% (1)
- Z1. Seminario 3 - 2023 - Unidad 3 - Condiciones de Vida y Su Relación Con La SaludDocumento70 páginasZ1. Seminario 3 - 2023 - Unidad 3 - Condiciones de Vida y Su Relación Con La SaludNahuel Diaz RomeroAún no hay calificaciones
- DiagnosticoFinalLaEncantadaMilagros PDFDocumento23 páginasDiagnosticoFinalLaEncantadaMilagros PDFJulio MTAún no hay calificaciones
- Semana 3. Métodos de Eliminación de MicroorganismosDocumento24 páginasSemana 3. Métodos de Eliminación de Microorganismosmarcia paguay100% (1)
- 17 02 Pie ReumáticoDocumento21 páginas17 02 Pie ReumáticoFabiola Fuentes RomeroAún no hay calificaciones
- Course Navette 1º BtoDocumento4 páginasCourse Navette 1º BtojavierenriquehoyossanchezAún no hay calificaciones
- Copia de Psicologia ClinicaDocumento2 páginasCopia de Psicologia ClinicaElodia Altagracia Altagracia ValdezAún no hay calificaciones
- PLAN - DE - VIGILANCIA, - PREVENCION - Y - CONTROL - DE - COVID-19 Mega ObraDocumento25 páginasPLAN - DE - VIGILANCIA, - PREVENCION - Y - CONTROL - DE - COVID-19 Mega ObraJose Povis EgoavilAún no hay calificaciones
- Tesis Argumentativa Un3 ArgDocumento4 páginasTesis Argumentativa Un3 ArgajenosAún no hay calificaciones
- Pae Lic Silvia EstradaDocumento17 páginasPae Lic Silvia EstradaVentura Gilber FGAún no hay calificaciones
- diagnostico Ptar hechoDocumento22 páginasdiagnostico Ptar hechoJhosé SsjAún no hay calificaciones
- Manual Mascotas El DoradoDocumento6 páginasManual Mascotas El Doradodoaguirre100% (1)
- Sopa Letras Taller Seguridad y Cultura Vial.....Documento1 páginaSopa Letras Taller Seguridad y Cultura Vial.....Luis David Mdr GmzAún no hay calificaciones
- Escala AUTOCONTROLDocumento129 páginasEscala AUTOCONTROLCEAún no hay calificaciones
- Estrategias para Promover La Actividad FísicaDocumento43 páginasEstrategias para Promover La Actividad FísicaCaRichardsWAún no hay calificaciones
- Rg-17-Sig-ymn - Inducción General de Seguridad y Salud en El TrabajoDocumento1 páginaRg-17-Sig-ymn - Inducción General de Seguridad y Salud en El TrabajoPame acAún no hay calificaciones
- ComentarioDocumento6 páginasComentarioFiorella Távara Puemape100% (2)
- Catálogo Perú Ciclo 5 - 2020 Navidad Virtual 1.0Documento52 páginasCatálogo Perú Ciclo 5 - 2020 Navidad Virtual 1.0AreliChilconTelloAún no hay calificaciones
- Modulo 2 Enfermedad Alergica Respiratoria.Documento32 páginasModulo 2 Enfermedad Alergica Respiratoria.Andrea PersiaAún no hay calificaciones
- DCM fMRIDocumento12 páginasDCM fMRIFelipe ÁlvarezAún no hay calificaciones