También podría gustarte
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Lab Trastornos HematologicosDocumento32 páginasLab Trastornos HematologicosNatalia PocasangreAún no hay calificaciones
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleAún no hay calificaciones
- Clase 1, Introducción a la Medicina Oral y Métodos de Estudio en Medicina Oral (1)Documento15 páginasClase 1, Introducción a la Medicina Oral y Métodos de Estudio en Medicina Oral (1)Cristian Pincheira ArayaAún no hay calificaciones
- Uso de Derivados del Cannabis en Pacientes con CáncerDe EverandUso de Derivados del Cannabis en Pacientes con CáncerAún no hay calificaciones
- Clasificacion AnemiasDocumento8 páginasClasificacion AnemiasmercedesAún no hay calificaciones
- Anemia HemolíticaDocumento62 páginasAnemia HemolíticajosueAún no hay calificaciones
- Lectura 1 Biometria Hematica PDFDocumento6 páginasLectura 1 Biometria Hematica PDFJose Juan Sobampo RochaAún no hay calificaciones
- Anemia hemolítica: clasificación y mecanismosDocumento15 páginasAnemia hemolítica: clasificación y mecanismosDavid BlancoAún no hay calificaciones
- Anemia de Padecimientos Cronicos 2024Documento10 páginasAnemia de Padecimientos Cronicos 2024kmh646tdmnAún no hay calificaciones
- Abordaje de AnemiasDocumento30 páginasAbordaje de AnemiasCeci SalazAún no hay calificaciones
- Biometría HemáticaDocumento30 páginasBiometría HemáticajavierAún no hay calificaciones
- Simposio: La Interpretación Del Hemograma.Documento54 páginasSimposio: La Interpretación Del Hemograma.Ivette RosarioAún no hay calificaciones
- Hemograma: Análisis de células sanguíneasDocumento6 páginasHemograma: Análisis de células sanguíneasMaira EspinozaAún no hay calificaciones
- Biometria HematicaDocumento8 páginasBiometria HematicaLayla MontesdeocaAún no hay calificaciones
- Sindrome AnemicoDocumento7 páginasSindrome AnemicoVanessa MendozaAún no hay calificaciones
- T.2 HematologiaDocumento30 páginasT.2 HematologiaRocio DuranteAún no hay calificaciones
- PDF 20230119 201952 0000Documento21 páginasPDF 20230119 201952 0000Karla JackeAún no hay calificaciones
- Anemia - Fisiología.Documento5 páginasAnemia - Fisiología.Camila Ñiquen ParejaAún no hay calificaciones
- Biometría Hemática: Instituto para El Desarrollo Y Actualizacion de PrefesionalesDocumento17 páginasBiometría Hemática: Instituto para El Desarrollo Y Actualizacion de PrefesionalesMiguel Ángel Roque HernándezAún no hay calificaciones
- Informe 06 AnemiaDocumento22 páginasInforme 06 AnemiaArelyAún no hay calificaciones
- Interpretacion BiometriaDocumento36 páginasInterpretacion BiometriaSeiko RodriguezAún no hay calificaciones
- Lectiras AnemiaDocumento10 páginasLectiras AnemiaNathaly MejíaAún no hay calificaciones
- Hematologia Cto ResumenesDocumento31 páginasHematologia Cto ResumenesRafael Acosta HernandezAún no hay calificaciones
- Tarea 5.1 Tejido Sanguíneo HISTO 1 DR LUIS PEREZ MENDEZDocumento10 páginasTarea 5.1 Tejido Sanguíneo HISTO 1 DR LUIS PEREZ MENDEZScarlin RodríguezAún no hay calificaciones
- Anemias - Dinámica de GrupoDocumento17 páginasAnemias - Dinámica de GrupoSteven ContrerasAún no hay calificaciones
- Anemia: causas, síntomas y tratamientoDocumento12 páginasAnemia: causas, síntomas y tratamientoJosé Parrilla Masías100% (1)
- ERITROGRAMADocumento27 páginasERITROGRAMACarlos Arturo Guzman50% (2)
- Anemia en EsDocumento16 páginasAnemia en EsCarlosAún no hay calificaciones
- Práctica N°10 FisiologíaDocumento5 páginasPráctica N°10 FisiologíaNadia Barbara MolloAún no hay calificaciones
- Anemia: causas, clasificación y manifestaciones clínicasDocumento40 páginasAnemia: causas, clasificación y manifestaciones clínicasestudiar123Aún no hay calificaciones
- Anemia FerropenicaDocumento6 páginasAnemia FerropenicaJosue GomezAún no hay calificaciones
- SX AnemicoDocumento2 páginasSX AnemicoSilvia Varela EspinozaAún no hay calificaciones
- Anemia For The Primary - En.es PDFDocumento16 páginasAnemia For The Primary - En.es PDFRugeles DanielAún no hay calificaciones
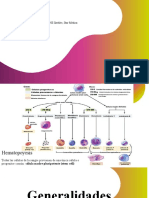
- GeneralidadesDocumento4 páginasGeneralidadesGabriel EspejoAún no hay calificaciones
- Anemia en PediatriaDocumento41 páginasAnemia en PediatriaBismark S. ChévezAún no hay calificaciones
- Síndrome Anemico (Guía)Documento9 páginasSíndrome Anemico (Guía)Edixon GarciaAún no hay calificaciones
- Exámenes de LaboratorioDocumento33 páginasExámenes de LaboratorioDiana Badillo100% (1)
- Valores Normales de Laboratorio Grupo 2Documento48 páginasValores Normales de Laboratorio Grupo 2ashley benitezAún no hay calificaciones
- Clasificacion de Las Anemias - Formacion IntegralDocumento14 páginasClasificacion de Las Anemias - Formacion IntegralIvana Daniela BelloAún no hay calificaciones
- Guía de Fisiología IifaseDocumento14 páginasGuía de Fisiología Iifaseyaffet chaconAún no hay calificaciones
- Seminario 6Documento19 páginasSeminario 6Romy PechAún no hay calificaciones
- Enfermedades hematológicas: anemia y eritrocitosisDocumento36 páginasEnfermedades hematológicas: anemia y eritrocitosisGenessisDayana HuertacruzAún no hay calificaciones
- TEMA II Sintesis y Catabolismo Del Hem y La GlobinaDocumento5 páginasTEMA II Sintesis y Catabolismo Del Hem y La Globinakaren campañaAún no hay calificaciones
- Marco Teorico AnemiaDocumento6 páginasMarco Teorico AnemiabeberlyAún no hay calificaciones
- Anemia SemioDocumento20 páginasAnemia SemioARMAS JOSSELYN NAZARETHAún no hay calificaciones
- Las AnemiasDocumento25 páginasLas AnemiasC&R DIGITAL COMPUTERSAún no hay calificaciones
- Anemia AbordajeDocumento5 páginasAnemia AbordajeCora OlaldesAún no hay calificaciones
- Resumen. Introducción Al Estudio de La AnemiaDocumento6 páginasResumen. Introducción Al Estudio de La AnemiaDiana Isabel De Los Santos PinedaAún no hay calificaciones
- 2017 - CNA Anemia - En.esDocumento16 páginas2017 - CNA Anemia - En.esSandra Milena Roberto HernandezAún no hay calificaciones
- Anemia de Los ParásitosDocumento18 páginasAnemia de Los ParásitosVirnia Patzi100% (1)
- Policitemia VeraDocumento8 páginasPolicitemia VeraDenis Hinojosa VelaAún no hay calificaciones
- La AnemiaDocumento12 páginasLa AnemiaCinthya BenavidesAún no hay calificaciones
- Celulas Sanguine As y HemogramaDocumento47 páginasCelulas Sanguine As y Hemogramanadiesta100% (1)
- Anemia FichaDocumento2 páginasAnemia Fichawilmary cuellarAún no hay calificaciones
- INTRODUCCIÓNDocumento8 páginasINTRODUCCIÓNPilar Carranza GrausAún no hay calificaciones
- Anemia Hemolítica y Anemia Consecutiva A Hemorragia AgudaDocumento4 páginasAnemia Hemolítica y Anemia Consecutiva A Hemorragia AgudaLoren Flórez MAún no hay calificaciones
- Seminario Sanguineo - MonografiaDocumento23 páginasSeminario Sanguineo - MonografiaJesus DnielAún no hay calificaciones
- Exámenes de LaboratorioDocumento30 páginasExámenes de LaboratorioMatias HidalgoAún no hay calificaciones
- Anemia: clasificación, causas y diagnósticoDocumento39 páginasAnemia: clasificación, causas y diagnósticodenisseAún no hay calificaciones
- CamposDocumento2 páginasCamposCan cocom Andrea auroraAún no hay calificaciones
- INTERLEUCINADocumento2 páginasINTERLEUCINACan cocom Andrea auroraAún no hay calificaciones
- Manual ortográfico universitarioDocumento24 páginasManual ortográfico universitarioCan cocom Andrea auroraAún no hay calificaciones
- Barrera HemaDocumento1 páginaBarrera HemaCan cocom Andrea auroraAún no hay calificaciones
- MeningesDocumento1 páginaMeningesCan cocom Andrea auroraAún no hay calificaciones
- UNEMEDocumento14 páginasUNEMECan cocom Andrea auroraAún no hay calificaciones
- Proyecto de Proceso EmfermeroDocumento13 páginasProyecto de Proceso EmfermeroCan cocom Andrea auroraAún no hay calificaciones
- La isoinmunización Rh: causas y consecuencias de la enfermedad hemolítica perinatalDocumento64 páginasLa isoinmunización Rh: causas y consecuencias de la enfermedad hemolítica perinatalNoe VGAún no hay calificaciones
- Metodos de CoagulacionDocumento16 páginasMetodos de CoagulacionDanielRiosRoblesAún no hay calificaciones
- Inmunohematologia EritrocitariaDocumento2 páginasInmunohematologia EritrocitariaCarlVDAún no hay calificaciones
- SangreDocumento4 páginasSangreaplata9Aún no hay calificaciones
- Hematología - Adriana PalaciosDocumento314 páginasHematología - Adriana PalaciosRubens UrbinaAún no hay calificaciones
- 01 Resultados SaludDignaDocumento7 páginas01 Resultados SaludDignaRosivel LópezAún no hay calificaciones
- Hematología Completa: Qué Es y Valores Normales: Tua Saúde Exámenes de DiagnósticoDocumento16 páginasHematología Completa: Qué Es y Valores Normales: Tua Saúde Exámenes de Diagnósticoyosman lópezAún no hay calificaciones
- Pruebas de Coagulacion: Tiempo de Control (10-14 Seg)Documento1 páginaPruebas de Coagulacion: Tiempo de Control (10-14 Seg)Ana Cecilia MantillaAún no hay calificaciones
- Jasell Cesar Ureña Gonzalez: Fecha de Entrada: 11-07-22 Página 1Documento6 páginasJasell Cesar Ureña Gonzalez: Fecha de Entrada: 11-07-22 Página 1Jasell CesarAún no hay calificaciones
- Sindrome AnemicoDocumento102 páginasSindrome AnemicoShammy LopezAún no hay calificaciones
- ExanguinotransfusiónDocumento10 páginasExanguinotransfusiónZitzzany MezaAún no hay calificaciones
- Áreas de Un Laboratorio ClínicoDocumento2 páginasÁreas de Un Laboratorio Clínicogioeuni7100% (3)
- TAH05 ContenidosDocumento8 páginasTAH05 ContenidosLucy ÑaaAún no hay calificaciones
- Laboratorio San RafaelDocumento2 páginasLaboratorio San RafaelBetina MartinAún no hay calificaciones
- Hemostasia: proceso de coagulación de la sangreDocumento18 páginasHemostasia: proceso de coagulación de la sangreFacuperrAún no hay calificaciones
- Interpretacion Del HemogramaDocumento46 páginasInterpretacion Del HemogramaSara Soria EstrugoAún no hay calificaciones
- Patologia Quiz HematologíaDocumento4 páginasPatologia Quiz HematologíadoctortritoAún no hay calificaciones
- Valores Bioquímicos de ReferenciaDocumento1 páginaValores Bioquímicos de ReferenciaMaria VallejosAún no hay calificaciones
- 1 - Historia ClínicaDocumento13 páginas1 - Historia ClínicaDiego AlvarezAún no hay calificaciones
- 2 Banco de Sangré - 20231211 - 164437 - 0000Documento7 páginas2 Banco de Sangré - 20231211 - 164437 - 0000Abanto Dagiro MartinAún no hay calificaciones
- Result A Do Lab OratorioDocumento2 páginasResult A Do Lab OratorioNicolay JoyaAún no hay calificaciones
- Globulos Rojos AnormalesDocumento1 páginaGlobulos Rojos AnormalesLuis Alan100% (1)
- Hemograma y perfil bioquímico completoDocumento9 páginasHemograma y perfil bioquímico completoMarinaAún no hay calificaciones
- Control de Calidad de HemostasiaDocumento52 páginasControl de Calidad de HemostasiaHugo Huaman MuñozAún no hay calificaciones
- Licon Inserto TechnolabDocumento4 páginasLicon Inserto TechnolabMarco LandaAún no hay calificaciones
- Grupo Sanguíneo y Factor RHDocumento6 páginasGrupo Sanguíneo y Factor RHGustavo RodriguezAún no hay calificaciones
- Memorias de Un ProfesionalDocumento7 páginasMemorias de Un Profesionalbrisney15Aún no hay calificaciones
- Hematología 2023Documento65 páginasHematología 2023Alejandro NuñezAún no hay calificaciones
- J Pruebas de CompatibilidadDocumento38 páginasJ Pruebas de CompatibilidadFernando Ardaya ReyesAún no hay calificaciones
- FISIOLOGIADocumento33 páginasFISIOLOGIALuis Octavio VillanuevaAún no hay calificaciones
- Fisiopatología de las enfermedades cardiovascularesDe EverandFisiopatología de las enfermedades cardiovascularesCalificación: 5 de 5 estrellas5/5 (1)
- Terapia cognitiva: Conceptos básicos y profundizaciónDe EverandTerapia cognitiva: Conceptos básicos y profundizaciónCalificación: 5 de 5 estrellas5/5 (1)
- TDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosDe EverandTDAH en Adultos. Cómo Reconocer y Tratar a un Adulto con TDAH en 30 Fáciles PasosCalificación: 4 de 5 estrellas4/5 (8)
- Psiconeuroinmunología para la práctica clínicaDe EverandPsiconeuroinmunología para la práctica clínicaCalificación: 5 de 5 estrellas5/5 (4)
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- Ansiedad infantil. Los trastornos explicados a los padresDe EverandAnsiedad infantil. Los trastornos explicados a los padresCalificación: 4.5 de 5 estrellas4.5/5 (25)
- El libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)De EverandEl libro conciso de la punción seca: Manual del terapeuta para las aplicaciones en los puntos gatillo miofasciales (Color)Calificación: 3 de 5 estrellas3/5 (2)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- Genética general: Libro de textoDe EverandGenética general: Libro de textoCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Trauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesDe EverandTrauma, miedo y amor: Hacia una autonomía interior con la ayuda de las constelacionesCalificación: 1 de 5 estrellas1/5 (1)
- Manual para la administración de medicamentos desde el proceso de atención de enfermería: Un enfoque para la seguridad del pacienteDe EverandManual para la administración de medicamentos desde el proceso de atención de enfermería: Un enfoque para la seguridad del pacienteCalificación: 2.5 de 5 estrellas2.5/5 (4)
- Puntos gatillo y puntos acupunturales (Color)De EverandPuntos gatillo y puntos acupunturales (Color)Calificación: 4.5 de 5 estrellas4.5/5 (13)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- El autismo: Reflexiones y pautas para comprenderlo y abordarloDe EverandEl autismo: Reflexiones y pautas para comprenderlo y abordarloCalificación: 4 de 5 estrellas4/5 (7)
- El concepto Mulligan de terapia manual (Color)De EverandEl concepto Mulligan de terapia manual (Color)Calificación: 5 de 5 estrellas5/5 (3)
- Póngase En Forma Sin Salir De Su CasaDe EverandPóngase En Forma Sin Salir De Su CasaCalificación: 4.5 de 5 estrellas4.5/5 (4)
- Prescripción de ejercico físico para la saludDe EverandPrescripción de ejercico físico para la saludCalificación: 5 de 5 estrellas5/5 (1)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Fundamentos de medicina tradicional chinaDe EverandFundamentos de medicina tradicional chinaCalificación: 4.5 de 5 estrellas4.5/5 (5)
- Terapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaDe EverandTerapia de vidas pasadas: Un camino hacia la luz del alma. Técnica y prácticaCalificación: 4.5 de 5 estrellas4.5/5 (11)
- Puntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)De EverandPuntos gatillo y cadenas musculares funcionales en osteopatía y terapia manual (Bicolor)Calificación: 4.5 de 5 estrellas4.5/5 (23)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)