También podría gustarte
- Kit de Actividades Infantiles: Material Con Base BíblicaDocumento28 páginasKit de Actividades Infantiles: Material Con Base BíblicaLuz Maria Castro BarreraAún no hay calificaciones
- Banco de PreguntasDocumento23 páginasBanco de PreguntasKRISTY LILIBETH LOPEZ DELGADOAún no hay calificaciones
- El Papel de La Mujer en La Revolución MexicanaDocumento17 páginasEl Papel de La Mujer en La Revolución MexicanaJosé Luis Cervantes CortésAún no hay calificaciones
- Inmunodeficiencias CongénitasDocumento36 páginasInmunodeficiencias CongénitasmisscinderellaAún no hay calificaciones
- InmunodeficienciasDocumento79 páginasInmunodeficienciasNaz Zegarra BellinaAún no hay calificaciones
- Preguntas Examen InvertebradosDocumento5 páginasPreguntas Examen Invertebradosadriana884100% (1)
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleDe EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleAún no hay calificaciones
- Inmunodeficiencias Primarias y SecundariasDocumento46 páginasInmunodeficiencias Primarias y Secundariasstephani hernandezAún no hay calificaciones
- Los cambios vasculares en la fisiopatología de las enfermedades en pequeños animales: Las nuevas teorías neurocirculatorias desde un enfoque clínicoDe EverandLos cambios vasculares en la fisiopatología de las enfermedades en pequeños animales: Las nuevas teorías neurocirculatorias desde un enfoque clínicoAún no hay calificaciones
- Errores innatos del metabolismo en el recién nacido: Abordaje clínicoDe EverandErrores innatos del metabolismo en el recién nacido: Abordaje clínicoAún no hay calificaciones
- Que Nos Enseña El PaseDocumento3 páginasQue Nos Enseña El PaseJulio RiverosAún no hay calificaciones
- Deficiencias de Los Componentes de Las Vías de Activación Clásica y Alternativa Del Sistema Del ComplementoDocumento6 páginasDeficiencias de Los Componentes de Las Vías de Activación Clásica y Alternativa Del Sistema Del ComplementoJesus Aguirre ReyesAún no hay calificaciones
- Tsa2 5497 29-05-2021Documento10 páginasTsa2 5497 29-05-2021ATILIO ALEJANDRO ALVAREZ RAMOSAún no hay calificaciones
- MicrobiologíaDocumento2 páginasMicrobiologíadaniela ojeda otaloraAún no hay calificaciones
- Defiencia Complemento 1Documento5 páginasDefiencia Complemento 1JOSELINE BELEN PEÑA FUENTESAún no hay calificaciones
- Inmunodeficiencias Primarias. Innatas. Deficiencias Del Sistema ComplementoDocumento7 páginasInmunodeficiencias Primarias. Innatas. Deficiencias Del Sistema ComplementoLeoAún no hay calificaciones
- Apuntes InmunodeficienciasDocumento7 páginasApuntes InmunodeficienciasAlicia Arroyo NogalesAún no hay calificaciones
- Tasca 4Documento4 páginasTasca 4Alexandra De SalinasAún no hay calificaciones
- 04 Angioedema C1INHDocumento19 páginas04 Angioedema C1INHNora MirandaAún no hay calificaciones
- Complemento Mapa ConceptualDocumento14 páginasComplemento Mapa ConceptualJDominguezBernalAún no hay calificaciones
- INMUNODEFICIENCIASDocumento39 páginasINMUNODEFICIENCIASSha Uxua Espinoza Zambrano100% (1)
- Inmunodeficiencias ClaseDocumento18 páginasInmunodeficiencias ClaseGESU PIERO LOPEZ MEJIAAún no hay calificaciones
- Sistema Del Complemento - Inmunología y Trastornos Alérgicos - Manual MSD Versión para ProfesionalesDocumento2 páginasSistema Del Complemento - Inmunología y Trastornos Alérgicos - Manual MSD Versión para ProfesionalesWilliam Aarón Vargas SalmerónAún no hay calificaciones
- Funciones Biológicas de Las Proteínas Del Complemento y DeficienciasDocumento8 páginasFunciones Biológicas de Las Proteínas Del Complemento y DeficienciasSandra VilloAún no hay calificaciones
- Inmunodeficiencias Congénitas y AdquiridasDocumento4 páginasInmunodeficiencias Congénitas y AdquiridasAlbertAún no hay calificaciones
- Clase 8 - INMUNODEFICIENCIASDocumento3 páginasClase 8 - INMUNODEFICIENCIASMaria PerezAún no hay calificaciones
- Seminario 8 - INMUNODEFICIENCIADocumento22 páginasSeminario 8 - INMUNODEFICIENCIALEYLA LUZ ESTRELLA BARNARD LAGUNAAún no hay calificaciones
- GUÍA CLASE 22 2023-1 - Inmunodef Prim y SecundDocumento10 páginasGUÍA CLASE 22 2023-1 - Inmunodef Prim y SecundVale AlvarezAún no hay calificaciones
- Caso Clinico InmunoDocumento30 páginasCaso Clinico InmunoFranciscoRiveraHernándezAún no hay calificaciones
- Temas de MonografíasDocumento16 páginasTemas de MonografíasLeila Natalia MARTINICAún no hay calificaciones
- Trabajo de Investigacion BioquimicaDocumento11 páginasTrabajo de Investigacion BioquimicaWesley GodoyAún no hay calificaciones
- Cap 18Documento5 páginasCap 18Jannet RVAún no hay calificaciones
- Inmunodeficiencias Congénitas (Primarias) : Afecta A La Vía Del Complemento o A Los FagocitosDocumento5 páginasInmunodeficiencias Congénitas (Primarias) : Afecta A La Vía Del Complemento o A Los FagocitosKarla Alejandra BriseñoAún no hay calificaciones
- Defectos de Los FagocitosDocumento14 páginasDefectos de Los Fagocitosjaguar8008Aún no hay calificaciones
- Sistema de Complemento y DeficienciaDocumento37 páginasSistema de Complemento y Deficienciajheimy100% (1)
- Defectos en La Fagocitosis. Aspectos Clínicos, Moleculares y TerapéuticosDocumento13 páginasDefectos en La Fagocitosis. Aspectos Clínicos, Moleculares y Terapéuticoscandy lmAún no hay calificaciones
- Trabajo Inmuno Upao 3 CicloDocumento23 páginasTrabajo Inmuno Upao 3 CicloNastia OlenkaAún no hay calificaciones
- Montenegro, CeciliaDocumento25 páginasMontenegro, CeciliaEleonor Talavera VargasAún no hay calificaciones
- El Sistema de ComplementoDocumento22 páginasEl Sistema de ComplementoElianethDelAngel100% (1)
- Enfermedades Por InmunodeficienciaDocumento2 páginasEnfermedades Por InmunodeficienciaEstefani Abigail Bollates SantosAún no hay calificaciones
- Lectura InmunodeficienciasDocumento7 páginasLectura InmunodeficienciasLuisa Fernanda Pedraza NarváezAún no hay calificaciones
- Enfermedades Autoinmunes Primarias y SecundariasDocumento17 páginasEnfermedades Autoinmunes Primarias y SecundariasLuis RodríguezAún no hay calificaciones
- 17 Inmunodeficiencias NNPDocumento32 páginas17 Inmunodeficiencias NNPGilbert CanedoAún no hay calificaciones
- ExpooDocumento3 páginasExpooJeison CuencaAún no hay calificaciones
- Estados de Inmunodeficiencia Linfocitos T y BDocumento19 páginasEstados de Inmunodeficiencia Linfocitos T y BFabiana MoraoAún no hay calificaciones
- Anemia Hemolítica CongénitaDocumento4 páginasAnemia Hemolítica CongénitaJenny ReascosAún no hay calificaciones
- Guía Práctica de Complemento Septiembre 2011Documento7 páginasGuía Práctica de Complemento Septiembre 2011InmunoBlog100% (1)
- INTRODUCCIONDocumento2 páginasINTRODUCCIONf5hdjbwb5bAún no hay calificaciones
- Capitulo 18 ReguerioDocumento2 páginasCapitulo 18 Reguerioanaca59505Aún no hay calificaciones
- Preguntas Inmunologìa y Genètica PDFDocumento5 páginasPreguntas Inmunologìa y Genètica PDFLi SacAún no hay calificaciones
- Inmunodeficiencia Primaria IdpDocumento16 páginasInmunodeficiencia Primaria IdpEmilia AcostaAún no hay calificaciones
- Deficiencia de Adhesión Leucocitaria Tipo 1Documento5 páginasDeficiencia de Adhesión Leucocitaria Tipo 1megajhonnyAún no hay calificaciones
- Hipersensibilidad Tipo Iii y IvDocumento73 páginasHipersensibilidad Tipo Iii y IvAnthony Klein Núñez AlarcónAún no hay calificaciones
- Inmunodeficiencias Primarias. Clínica y Formas VariantesDocumento25 páginasInmunodeficiencias Primarias. Clínica y Formas VariantesSergio VasquezAún no hay calificaciones
- Gen Del LupusDocumento2 páginasGen Del LupusPabloAún no hay calificaciones
- El Sistema Del ComplementoDocumento8 páginasEl Sistema Del ComplementoFabiana MoraoAún no hay calificaciones
- Clase 1 LoretoDocumento3 páginasClase 1 LoretoDavid AlvaradoAún no hay calificaciones
- Inmunodeficiencia Adquirida de Origen VíricoDocumento25 páginasInmunodeficiencia Adquirida de Origen VíricoMercedes MartínezAún no hay calificaciones
- .Sistema de Complemento .Documento11 páginas.Sistema de Complemento .Janacua Figueroa DianaAún no hay calificaciones
- Inmunodeficiencias Primarias y SecundarriasDocumento36 páginasInmunodeficiencias Primarias y SecundarriasKevin Josué Campos GuzmánAún no hay calificaciones
- La Regulación Del Sistema Del ComplementoDocumento3 páginasLa Regulación Del Sistema Del ComplementoTuyaPuscànSayuriAún no hay calificaciones
- INMUNODEFICIENCIADocumento4 páginasINMUNODEFICIENCIAAndrea BorreroAún no hay calificaciones
- Inmunodeficiencias Primarias y SecundariasDocumento14 páginasInmunodeficiencias Primarias y SecundariasJandry MurilloAún no hay calificaciones
- 113-Texto Del Artículo, Manuscrito o Aportación A La Revista-309-1!11!20201223Documento17 páginas113-Texto Del Artículo, Manuscrito o Aportación A La Revista-309-1!11!20201223Julia VargasAún no hay calificaciones
- Anato ESPACIOS PERIFARINGEOSDocumento5 páginasAnato ESPACIOS PERIFARINGEOSJulia VargasAún no hay calificaciones
- Ojo Retinopatia DiabéticaDocumento1 páginaOjo Retinopatia DiabéticaJulia VargasAún no hay calificaciones
- Nariz, Boca y Glándulas SalivalesDocumento15 páginasNariz, Boca y Glándulas SalivalesJulia VargasAún no hay calificaciones
- Ecografía Básica - Repaso FinalDocumento52 páginasEcografía Básica - Repaso FinalJulia VargasAún no hay calificaciones
- Grupo 03-Shock HipovolemicoDocumento26 páginasGrupo 03-Shock HipovolemicoJulia VargasAún no hay calificaciones
- Historia Clinica Vargas Ventura - Vasquez JaramilloDocumento12 páginasHistoria Clinica Vargas Ventura - Vasquez JaramilloJulia VargasAún no hay calificaciones
- Caso Clinico NeumoníaDocumento2 páginasCaso Clinico NeumoníaJulia VargasAún no hay calificaciones
- Discapacidad Intelectual Infantil Investigacion Cualitativa 2Documento18 páginasDiscapacidad Intelectual Infantil Investigacion Cualitativa 2teresa margaritaAún no hay calificaciones
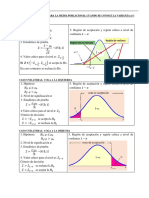
- Prueba de Hipotesis para La Media Con Varianza ConocidaDocumento2 páginasPrueba de Hipotesis para La Media Con Varianza Conocidajudit cristina ramirez floresAún no hay calificaciones
- Informe Lab1 Electronica 1Documento8 páginasInforme Lab1 Electronica 1Brian SanchezAún no hay calificaciones
- 63 Afirmaciones para Ser IrresistibleDocumento3 páginas63 Afirmaciones para Ser IrresistibleCristiánAún no hay calificaciones
- Problema para Muestra de Matemáticas 17-18Documento1 páginaProblema para Muestra de Matemáticas 17-18Ana ValleAún no hay calificaciones
- Grado Sexto Guia 1 - Logica y Conjuntos (Tema1Proposiciones)Documento7 páginasGrado Sexto Guia 1 - Logica y Conjuntos (Tema1Proposiciones)Noel VelasquezAún no hay calificaciones
- Sara ChapotDocumento9 páginasSara ChapotnerofeAún no hay calificaciones
- Revista Digital - Fundación y VeterinariaDocumento4 páginasRevista Digital - Fundación y VeterinariaNazh VictoriaAún no hay calificaciones
- El Perdón Es en El Cristianismo El Acto de Amor Más ImportanteDocumento2 páginasEl Perdón Es en El Cristianismo El Acto de Amor Más ImportanteWalter CostasAún no hay calificaciones
- Lineas de Tiempo Calidad 5enDocumento13 páginasLineas de Tiempo Calidad 5enKamilo Andres Galindo ZanchesAún no hay calificaciones
- Guia EscrituraDocumento17 páginasGuia EscrituraAlvaro Moreno AlonsoAún no hay calificaciones
- Lesiones ReactivasDocumento13 páginasLesiones ReactivasAndreina ochoaAún no hay calificaciones
- Plan de Contingencia Distrital Feriado CarnavalDocumento11 páginasPlan de Contingencia Distrital Feriado CarnavalDeici CárdenazAún no hay calificaciones
- Resumen Grupos Unidad 4Documento16 páginasResumen Grupos Unidad 4Mauro Nicolás FernándezAún no hay calificaciones
- Operatoria Dental III 2016 PDFDocumento20 páginasOperatoria Dental III 2016 PDFFrankAquinoOrtizAún no hay calificaciones
- Presentación Signos de PuntuaciónDocumento7 páginasPresentación Signos de PuntuaciónAmparo Dussán VelásquezAún no hay calificaciones
- Intoxicación Por MetanolDocumento22 páginasIntoxicación Por MetanolJose QuinteroAún no hay calificaciones
- L.meso Aesthetic de EspañaDocumento19 páginasL.meso Aesthetic de EspañaOscar BelmanAún no hay calificaciones
- La Irrevocabilidad Del Reconocimiento de Hijo Extramatrimonial Vs Interés Superior Del Menor: A Propósito de La Consulta #132-2010-La LibertadDocumento15 páginasLa Irrevocabilidad Del Reconocimiento de Hijo Extramatrimonial Vs Interés Superior Del Menor: A Propósito de La Consulta #132-2010-La LibertadFranklin Silva YarangaAún no hay calificaciones
- Cronograma de Actividades Ese Hospital San MartinDocumento13 páginasCronograma de Actividades Ese Hospital San Martinelena anguloAún no hay calificaciones
- Cuadro La Independencia de MexicoDocumento5 páginasCuadro La Independencia de Mexicoleam1973Aún no hay calificaciones
- Modelo de Acta de ProyectoDocumento9 páginasModelo de Acta de ProyectoKapo Sur SurAún no hay calificaciones
- Lab 04 Transformada Z InversaDocumento19 páginasLab 04 Transformada Z InversaLuis RondoAún no hay calificaciones
- Tunel Del Viento Vs CFDDocumento8 páginasTunel Del Viento Vs CFDgoyoamagueAún no hay calificaciones
- QR 23 RRDocumento3 páginasQR 23 RRisabelacevedoAún no hay calificaciones
- Siddur 03 Rosh ChodeshDocumento13 páginasSiddur 03 Rosh ChodeshAvi Jew GAFC 7Aún no hay calificaciones