También podría gustarte
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Guía práctica para técnico superior de laboratorio de diagnóstico clínico y biomédicoDe EverandGuía práctica para técnico superior de laboratorio de diagnóstico clínico y biomédicoCalificación: 4.5 de 5 estrellas4.5/5 (10)
- Banco de Preguntas NefroDocumento26 páginasBanco de Preguntas Nefrochicho1chi75% (12)
- Hemato InfectoDocumento9 páginasHemato InfectoGianpiero Fernando Mas NicovaniAún no hay calificaciones
- Caso ClínicoDocumento33 páginasCaso ClínicoHéctor Alfonso Cuevas CruzAún no hay calificaciones
- Manual de patología clínica veterinariaDe EverandManual de patología clínica veterinariaCalificación: 4 de 5 estrellas4/5 (4)
- Analisis de Biometria HematicaDocumento4 páginasAnalisis de Biometria HematicaFer VacaAún no hay calificaciones
- Mir 01 1516 Desgloses HM Web PDFDocumento40 páginasMir 01 1516 Desgloses HM Web PDFAlex Medina Aguilar100% (5)
- Alteraciones de Los Eritrocitos PDFDocumento24 páginasAlteraciones de Los Eritrocitos PDFsandra maribel sucasaca suañaAún no hay calificaciones
- Alteraciones de Los Eritrocitos PDFDocumento24 páginasAlteraciones de Los Eritrocitos PDFsandra maribel sucasaca suañaAún no hay calificaciones
- Examen CENDEISSS Microbiología 2017Documento13 páginasExamen CENDEISSS Microbiología 2017David CantilloAún no hay calificaciones
- Caso Clinico AnemiaDocumento16 páginasCaso Clinico AnemiaYvon Becerra Cueva100% (1)
- ACTIVIDAD Trabajo Individual Primera Parte Comprensión EDocumento12 páginasACTIVIDAD Trabajo Individual Primera Parte Comprensión EIsabella Diaz OrtizAún no hay calificaciones
- Casos Clinicos para EnviarDocumento5 páginasCasos Clinicos para EnviarPatricia DonairesAún no hay calificaciones
- Gonzalez, Silvia Analía - AteneoDocumento10 páginasGonzalez, Silvia Analía - AteneoSilvia Analía GonzálezAún no hay calificaciones
- Actualizaciones HM 11Documento2 páginasActualizaciones HM 11lalobitaAún no hay calificaciones
- Ev de Aprendizaje 6 Problemas Basados en ProblemasDocumento20 páginasEv de Aprendizaje 6 Problemas Basados en Problemasbetto alvarezAún no hay calificaciones
- Patología RenalDocumento5 páginasPatología RenalTony Olivera BarzolaAún no hay calificaciones
- Guia Aaa de Practicas de Hematologia II 2013aDocumento61 páginasGuia Aaa de Practicas de Hematologia II 2013aErick Antonio P. GalindoAún no hay calificaciones
- Taller de Hematología 2019-1Documento4 páginasTaller de Hematología 2019-1Nancy MuñozAún no hay calificaciones
- Casos Clínicos Leucemias 2022Documento7 páginasCasos Clínicos Leucemias 2022Brandon MendozaAún no hay calificaciones
- Preguntas de Control ViiDocumento2 páginasPreguntas de Control ViiHellora PassafaroAún no hay calificaciones
- Caso ClínicoDocumento6 páginasCaso ClínicoVeronica Di GiannantonioAún no hay calificaciones
- Quipo 4 L5Documento3 páginasQuipo 4 L5Andrea IbarraAún no hay calificaciones
- REMV-Caso 2-Ev. 6Documento7 páginasREMV-Caso 2-Ev. 6Rubi VazAún no hay calificaciones
- Casos Clínicos 2do Exmn de PatologiaDocumento4 páginasCasos Clínicos 2do Exmn de PatologiaJohnny NestorAún no hay calificaciones
- Congreso Argentino de Hematología - Vol17Documento56 páginasCongreso Argentino de Hematología - Vol17emmassr7713Aún no hay calificaciones
- Informe #3 - Hemoglobina y HematocritoDocumento14 páginasInforme #3 - Hemoglobina y HematocritoLuis PAún no hay calificaciones
- 3 - Eritrocito 2017Documento8 páginas3 - Eritrocito 2017Camila Da Silva100% (1)
- Primer Parcial Patologia Clínica 2-2018Documento2 páginasPrimer Parcial Patologia Clínica 2-2018samuel naAún no hay calificaciones
- S E M I N A R I O: Interpretacion de HemogramasDocumento3 páginasS E M I N A R I O: Interpretacion de HemogramasJesus ParragaAún no hay calificaciones
- Parcial IDocumento8 páginasParcial IAngela Betancur FajardoAún no hay calificaciones
- Grupo 1Documento12 páginasGrupo 1Juan David Gomez0% (1)
- EnarmDocumento22 páginasEnarmAnwar GutierrezAún no hay calificaciones
- Recopilado UNINORTE 2021Documento10 páginasRecopilado UNINORTE 2021Julio Sergio BarriosAún no hay calificaciones
- 3era SemanaDocumento4 páginas3era SemanamierdactmAún no hay calificaciones
- Preguntas D EcografiaDocumento7 páginasPreguntas D EcografiaTP LissethAún no hay calificaciones
- Casos ClinicosDocumento12 páginasCasos ClinicosSilvia Eche echeAún no hay calificaciones
- Caso Clínico 25Documento2 páginasCaso Clínico 25Juan Domingo Davila GilAún no hay calificaciones
- Caso ClínicoDocumento5 páginasCaso ClínicoRhoy QRAún no hay calificaciones
- Determinación de Hemoglobina y Hematocrito.Documento5 páginasDeterminación de Hemoglobina y Hematocrito.Sheila M DiazAún no hay calificaciones
- Casos para He UapDocumento6 páginasCasos para He UapPaola CBAún no hay calificaciones
- Informe de Internado Oblitas BancoDocumento11 páginasInforme de Internado Oblitas BancoYesi Oblitas0% (1)
- Historia Clínica - GastroenterologíaDocumento4 páginasHistoria Clínica - GastroenterologíaLuz DíazAún no hay calificaciones
- Casos Clinicos 2Documento2 páginasCasos Clinicos 2Pecr PecrAún no hay calificaciones
- Sobredosis Letal Accidental de Colchicina: Accidental Fatal Colchicine OverdoseDocumento3 páginasSobredosis Letal Accidental de Colchicina: Accidental Fatal Colchicine OverdoseCecilio Díaz GijónAún no hay calificaciones
- TP Lab SangreDocumento4 páginasTP Lab SangreHernán LindónAún no hay calificaciones
- Hematies#5 - ADA 1 Casos Clinicos LHC2022Documento7 páginasHematies#5 - ADA 1 Casos Clinicos LHC2022marysobAún no hay calificaciones
- Ma 00010 03Documento3 páginasMa 00010 03Carmen Garcia AAún no hay calificaciones
- Caso ClinicoDocumento8 páginasCaso ClinicoKarla OrozcoAún no hay calificaciones
- Historia Clinica 1.2 FINALDocumento7 páginasHistoria Clinica 1.2 FINALNelson David Maldonado P.Aún no hay calificaciones
- Metodos de DiagnosticoDocumento4 páginasMetodos de DiagnosticoVinicius SilvaAún no hay calificaciones
- Casos de HematologíaDocumento15 páginasCasos de HematologíaDaniel SalazarAún no hay calificaciones
- Reporte de CasoDocumento4 páginasReporte de CasoJeffrey Kevin Minchola VegaAún no hay calificaciones
- Bioquímica - Nutrición NormalDocumento3 páginasBioquímica - Nutrición NormalLourdes PelocAún no hay calificaciones
- Cambios DisplasicosDocumento17 páginasCambios DisplasicosClemencia Alvarez VillaAún no hay calificaciones
- Caso Clínico Hematología - AiqmDocumento2 páginasCaso Clínico Hematología - AiqmArlette Quevedo M.Aún no hay calificaciones
- Sangre y Sus ConstantesDocumento4 páginasSangre y Sus Constanteskirc KL 201Aún no hay calificaciones
- Lectura de Hemograma..Documento9 páginasLectura de Hemograma..Clarita GallardoAún no hay calificaciones
- 1 ExamenDocumento3 páginas1 ExamenJohnny NestorAún no hay calificaciones
- Sindromes HematológicsDocumento92 páginasSindromes HematológicsAnakaren Renée Game IbacacheAún no hay calificaciones
- Copia Hematocrito y HemoglobinaDocumento14 páginasCopia Hematocrito y HemoglobinaSTEFHANY PAOLA MENDOZA HERNANDEZAún no hay calificaciones
- Tarea Grupal AnemiasDocumento12 páginasTarea Grupal AnemiasGunterRolandoAún no hay calificaciones
- Hematopoyesis Resume 65854 Downloadable 2070459Documento24 páginasHematopoyesis Resume 65854 Downloadable 2070459Stella maris FOSSATTIAún no hay calificaciones
- Es WinpetteDocumento4 páginasEs WinpetteSitri MenesesAún no hay calificaciones
- DLMB - AR - Etapa 4 - FGyBDocumento12 páginasDLMB - AR - Etapa 4 - FGyBDiana MeléndezAún no hay calificaciones
- Marco Teorico de MicroDocumento3 páginasMarco Teorico de MicroMimi MartinezAún no hay calificaciones
- Pauta Corrección Evaluación Formativa NM4 BiologíaDocumento10 páginasPauta Corrección Evaluación Formativa NM4 BiologíaLucky GimenezAún no hay calificaciones
- 9 New Classification Zygomycota - Pdf.en - Es.cvDocumento14 páginas9 New Classification Zygomycota - Pdf.en - Es.cvJorge A. SilvaAún no hay calificaciones
- Morin Edgar El Metodo 2 La Vida de La Vida ProcesadoDocumento539 páginasMorin Edgar El Metodo 2 La Vida de La Vida ProcesadoCindy Katerine Torres ArdilaAún no hay calificaciones
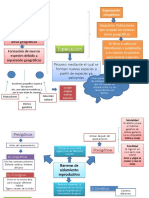
- Especiación y Barreras de Aislamiento ReproductivoDocumento2 páginasEspeciación y Barreras de Aislamiento ReproductivoAlan CovarrubiasAún no hay calificaciones
- Actividad Ciclo CelularDocumento8 páginasActividad Ciclo CelularSaid Guevara AriasAún no hay calificaciones
- La Evolución Del Ser HumanoDocumento2 páginasLa Evolución Del Ser HumanoJOSE LUISAún no hay calificaciones
- Alteraciones - Completo - Cari - Blancobrutal!!!! Apra Hacer Apuntes NuevosDocumento177 páginasAlteraciones - Completo - Cari - Blancobrutal!!!! Apra Hacer Apuntes NuevosAlicia de la CalAún no hay calificaciones
- Antropología Era BarceloDocumento13 páginasAntropología Era BarceloJuuAún no hay calificaciones
- Guía Biocel 3er PeriodoDocumento38 páginasGuía Biocel 3er PeriodoSof�a Victoria P�rez ContrerasAún no hay calificaciones
- Mapa Mixto BiologiaDocumento1 páginaMapa Mixto BiologiaSiranuch100% (2)
- LeccionDocumento1 páginaLeccionMAYKIN ESCRIBANO DELGADOAún no hay calificaciones
- Simposio de Creacionismo - Poder CreadorDocumento6 páginasSimposio de Creacionismo - Poder CreadorMis Huevadas XdAún no hay calificaciones
- La Danza de Las MoléculasDocumento16 páginasLa Danza de Las MoléculasOscar KissbertAún no hay calificaciones
- Infografia de La Celula Vegetal Maite Palacios Asto 215448 Downloable 3314487Documento2 páginasInfografia de La Celula Vegetal Maite Palacios Asto 215448 Downloable 3314487KELLY ANYELI PORTILLA FERNANDEZAún no hay calificaciones
- Jornalizacion de Contenidos Priorizados Quimica Ii y Quimica Iii BCHDocumento5 páginasJornalizacion de Contenidos Priorizados Quimica Ii y Quimica Iii BCHWendy QuirozAún no hay calificaciones
- Citoplasma PDFDocumento16 páginasCitoplasma PDFCatalina PobleteAún no hay calificaciones
- Er Examen Parcial Biologia 1 RespuestasDocumento3 páginasEr Examen Parcial Biologia 1 RespuestasMalena GonzálezAún no hay calificaciones
- Taller 2Documento3 páginasTaller 2BrunoAún no hay calificaciones
- Chable Et Al., 2017Documento31 páginasChable Et Al., 2017Jose Manuel Escutia PonceAún no hay calificaciones
- Celula IiDocumento2 páginasCelula IiCarmen ValeraAún no hay calificaciones
- Las Histonas y Su FunciònDocumento3 páginasLas Histonas y Su FunciònalfeterAún no hay calificaciones
- TAREA 1 de EvaluacionDocumento6 páginasTAREA 1 de EvaluacionDilenia MerejildoAún no hay calificaciones
- Tema 6 Sharon Mendoza SullaDocumento8 páginasTema 6 Sharon Mendoza SullaMily Gutierrez EspinozaAún no hay calificaciones
- NeurofisiologíaDocumento12 páginasNeurofisiologíaKarenshitAún no hay calificaciones
- Ciencias Naturales 6 A Zumbado 2021Documento20 páginasCiencias Naturales 6 A Zumbado 2021WendiiHernandezAún no hay calificaciones
- Clasificación de Los Ácidos NucleicosDocumento1 páginaClasificación de Los Ácidos NucleicosClases particularesAún no hay calificaciones
- Trabajo de Biologia ValeDocumento6 páginasTrabajo de Biologia ValeDaniel LopezAún no hay calificaciones