También podría gustarte
- UF1868 - Operación y supervisión de los equipos de conmutación telefónicaDe EverandUF1868 - Operación y supervisión de los equipos de conmutación telefónicaAún no hay calificaciones
- Validación farmacéutica: procesos, equipos, métodos y limpiezaDocumento28 páginasValidación farmacéutica: procesos, equipos, métodos y limpiezaAntonio DíazAún no hay calificaciones
- Sistema de mejora continua de la calidad en el laboratorio: Teoría y prácticaDe EverandSistema de mejora continua de la calidad en el laboratorio: Teoría y prácticaAún no hay calificaciones
- ValidacionDocumento131 páginasValidacionKarlos Martinez100% (1)
- Orientaciones para la evaluación de riesgos y las reglas de decisión según la Norma ISO/IEC 17025De EverandOrientaciones para la evaluación de riesgos y las reglas de decisión según la Norma ISO/IEC 17025Calificación: 4.5 de 5 estrellas4.5/5 (4)
- Validación Procesos ACFARMADocumento3 páginasValidación Procesos ACFARMAMeli Kieffer100% (6)
- Politica de Validacion - PMV - Doc de Validaciones y CalificacionesDocumento85 páginasPolitica de Validacion - PMV - Doc de Validaciones y Calificacionestito1628Aún no hay calificaciones
- Control de procesos y seguridad e higiene. INAV0109De EverandControl de procesos y seguridad e higiene. INAV0109Aún no hay calificaciones
- ValidacionDocumento131 páginasValidacionEdson Rafael Sánchez Monroy100% (3)
- El arte de probar: Guía sobre software testing para principiantes.De EverandEl arte de probar: Guía sobre software testing para principiantes.Aún no hay calificaciones
- Validaci N de Procesos GC2020Documento36 páginasValidaci N de Procesos GC2020Vicente Alejandro Arancibia AguileraAún no hay calificaciones
- UF1179 - Tratamientos previos de la lecheDe EverandUF1179 - Tratamientos previos de la lecheAún no hay calificaciones
- Guia ValidacionDocumento54 páginasGuia ValidacionAdriana Lopez100% (2)
- Gestión de la calidad ISO 9001/2015 en hosteleriaDe EverandGestión de la calidad ISO 9001/2015 en hosteleriaCalificación: 5 de 5 estrellas5/5 (1)
- Parcial 1 Valpro 2019Documento13 páginasParcial 1 Valpro 2019camilo andres gomez cabreraAún no hay calificaciones
- Guia de Inspeccion de Buenas Practicas D PDFDocumento55 páginasGuia de Inspeccion de Buenas Practicas D PDFRaúl Espinosa FabiánAún no hay calificaciones
- Gestión de la calidad (ISO 9001/2015)De EverandGestión de la calidad (ISO 9001/2015)Calificación: 3.5 de 5 estrellas3.5/5 (3)
- Propuesta GUIA de VALIDACIÓN IndustriaDocumento7 páginasPropuesta GUIA de VALIDACIÓN IndustriaAndy Rojas100% (1)
- Validacion Métodos Analíticos-FMDocumento88 páginasValidacion Métodos Analíticos-FMmanuelsnic2820100% (2)
- Validacion de Procesos-1 PDFDocumento50 páginasValidacion de Procesos-1 PDFCAMILAAún no hay calificaciones
- Auditoria Interna HaccpDocumento25 páginasAuditoria Interna HaccpGenaroPinosAún no hay calificaciones
- Validacion de ProcesosDocumento59 páginasValidacion de ProcesosEdward Saumeth100% (1)
- La Metrología en Los Sistemas de CalidadDocumento20 páginasLa Metrología en Los Sistemas de CalidadOmar Jair Purata SifuentesAún no hay calificaciones
- ASS-AYC-GU015 Validacion de ProcesosDocumento8 páginasASS-AYC-GU015 Validacion de Procesoszombiecorp100% (1)
- Exposición Validación Farmacéutica - Eq.2Documento40 páginasExposición Validación Farmacéutica - Eq.2preinscripcionesanaliticaAún no hay calificaciones
- Política de Equipos Críticos de Control de CalidadDocumento11 páginasPolítica de Equipos Críticos de Control de CalidadMariana AvilaAún no hay calificaciones
- Lista de Verificación VolumetriaDocumento6 páginasLista de Verificación Volumetriaeduardo hernandezAún no hay calificaciones
- Tarea 2Documento7 páginasTarea 2Logan PikmanAún no hay calificaciones
- Guia de Validación de Procesos 2019Documento8 páginasGuia de Validación de Procesos 2019Santiago CastrillonAún no hay calificaciones
- Traduccion Anexo 4 Informe 40Documento6 páginasTraduccion Anexo 4 Informe 40Vania Vargas QuisbertAún no hay calificaciones
- Validacion de Metodo Por UV de Furosemida - RemovedDocumento219 páginasValidacion de Metodo Por UV de Furosemida - Removedisis riosAún no hay calificaciones
- Recomendaciones de Manejo de Control Liofilizado AC 2016Documento12 páginasRecomendaciones de Manejo de Control Liofilizado AC 2016Patrick ThomasAún no hay calificaciones
- ValidaciónDocumento59 páginasValidaciónElsa María100% (3)
- 514 Revision Anual de ProductoDocumento5 páginas514 Revision Anual de Productochristian reckziegel0% (1)
- Tipos de ValidacionDocumento2 páginasTipos de ValidacionIsaac EstradaAún no hay calificaciones
- Tema 18 Requisitos Tecnicos Iso 17025Documento5 páginasTema 18 Requisitos Tecnicos Iso 17025monitriAún no hay calificaciones
- Nom 059 Ssa1 2013Documento97 páginasNom 059 Ssa1 2013Gustavo Rico0% (1)
- Validacion ProcesosDocumento59 páginasValidacion ProcesosHOSMANECHEVERRIA75% (4)
- Clase 13 Validacion de ProcesosDocumento68 páginasClase 13 Validacion de ProcesosAlexa BriAún no hay calificaciones
- Gestion de Equipos - Carreira PDFDocumento40 páginasGestion de Equipos - Carreira PDFJonathan CastelanAún no hay calificaciones
- Sesion 09 Validaciones Tecnologia 2023Documento92 páginasSesion 09 Validaciones Tecnologia 2023KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- Guia ValidacionDocumento55 páginasGuia ValidacionEdwin Alfonso Espinoza GutierrezAún no hay calificaciones
- Calificación de Equipos Segun BPLDocumento27 páginasCalificación de Equipos Segun BPLArmusickVentasBogAún no hay calificaciones
- Validación de Procesos (NOTAS)Documento15 páginasValidación de Procesos (NOTAS)camilo andres gomez cabreraAún no hay calificaciones
- Guia Validacion GMPDocumento56 páginasGuia Validacion GMPmaggie0batres1100% (3)
- FOR-LAB-009 Lista de Verificacion de Requisitos de GestionDocumento14 páginasFOR-LAB-009 Lista de Verificacion de Requisitos de GestionJosue Crispin RitoAún no hay calificaciones
- Informe 40 Español.Documento16 páginasInforme 40 Español.Guiss LemaAún no hay calificaciones
- Exigencias metrológicas ISO 9000 17025Documento21 páginasExigencias metrológicas ISO 9000 17025Nicolas RamosAún no hay calificaciones
- Cuestiones de Normas de Correctas FabricaciónDocumento3 páginasCuestiones de Normas de Correctas Fabricaciónandreangeles093Aún no hay calificaciones
- Guia ValidacionDocumento55 páginasGuia ValidacionFender Strat100% (1)
- Normas Iso 9002 y 9003Documento31 páginasNormas Iso 9002 y 9003Cristian Leiva83% (6)
- Actividad 6 Normas y Control de CalidadDocumento7 páginasActividad 6 Normas y Control de CalidadJuan Rivera AlcantaraAún no hay calificaciones
- ISO/TS 16949: 2009 Sistema Calidad AutomotrizDocumento19 páginasISO/TS 16949: 2009 Sistema Calidad AutomotrizAgustin Morales FloresAún no hay calificaciones
- Control de CalidadDocumento15 páginasControl de CalidadCarolyne LanzianoAún no hay calificaciones
- Validacion de ProveedoresDocumento35 páginasValidacion de ProveedoresCelio Barrigas RojasAún no hay calificaciones
- SGS CTS Procedure Reglas de Certificacion de Producto Letter ES 12 V5Documento32 páginasSGS CTS Procedure Reglas de Certificacion de Producto Letter ES 12 V5Tde Los RíosAún no hay calificaciones
- EquiposDocumento45 páginasEquiposRoli Lucana NaniAún no hay calificaciones
- 2-Validación de Métodos de Análisis Químico. Enfoque PrácticoDocumento124 páginas2-Validación de Métodos de Análisis Químico. Enfoque PrácticoVictor QuinterosAún no hay calificaciones
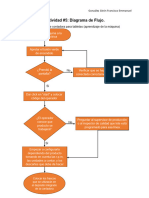
- Actividad #5: Diagrama de Flujo.: González Girón Francisco EmmanuelDocumento1 páginaActividad #5: Diagrama de Flujo.: González Girón Francisco EmmanuelPaco Gonzalez GironAún no hay calificaciones
- González Girón Francisco EmmanuelDocumento1 páginaGonzález Girón Francisco EmmanuelPaco Gonzalez GironAún no hay calificaciones
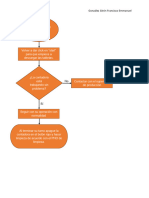
- Actividad #5: Diagrama de Flujo.: González Girón Francisco EmmanuelDocumento1 páginaActividad #5: Diagrama de Flujo.: González Girón Francisco EmmanuelPaco Gonzalez GironAún no hay calificaciones
- ÑoijliuhiiuDocumento6 páginasÑoijliuhiiuPaco Gonzalez GironAún no hay calificaciones
- Actividad 2 - Cuestionario Del Modulo 1 - Revisión Del IntentoDocumento3 páginasActividad 2 - Cuestionario Del Modulo 1 - Revisión Del IntentoAlejandro MedinaAún no hay calificaciones
- Errores de PronosticoDocumento2 páginasErrores de PronosticoKarla TinizarayAún no hay calificaciones
- Certificado de Calibración #Lc-0056-2022: Oscar F. Vivanco ValerioDocumento2 páginasCertificado de Calibración #Lc-0056-2022: Oscar F. Vivanco ValerioAntonio Segura FuentesAún no hay calificaciones
- Análisis de Sensibilidad ForoDocumento3 páginasAnálisis de Sensibilidad ForoDavid PerniaAún no hay calificaciones
- Responsabilidad Del Docente en La Formación Integral OrozcoDocumento47 páginasResponsabilidad Del Docente en La Formación Integral OrozcoedwinAún no hay calificaciones
- Optimización de Procesos ProductivosDocumento22 páginasOptimización de Procesos ProductivosDanilsonCortesLealAún no hay calificaciones
- Criterios para La Selección de Un Determinado Tipo de Estudio EpidemiologicoDocumento13 páginasCriterios para La Selección de Un Determinado Tipo de Estudio EpidemiologicoJorge Antonio YzRzAún no hay calificaciones
- TA OR080cDocumento7 páginasTA OR080cJorge CastilloAún no hay calificaciones
- Estándar Internacional: Gestión de Riesgos - Técnicas de Evaluación de RiesgosDocumento125 páginasEstándar Internacional: Gestión de Riesgos - Técnicas de Evaluación de RiesgosDiego Contreras HernándezAún no hay calificaciones
- Propiedades de Los Dispositivos de MedidaDocumento5 páginasPropiedades de Los Dispositivos de MedidajocelynAún no hay calificaciones
- Diapositiva Del Laboratorio 1 de Física 1 EPE - 2023 - 01Documento12 páginasDiapositiva Del Laboratorio 1 de Física 1 EPE - 2023 - 01Denisse RosalesAún no hay calificaciones
- Pasos para Determinar La Incertidumbre en Un Ensayo de LaboratorioDocumento10 páginasPasos para Determinar La Incertidumbre en Un Ensayo de Laboratoriojavier tamayoAún no hay calificaciones
- 5 Decisiones21Documento31 páginas5 Decisiones21Gabriel LeguaAún no hay calificaciones
- Finanzas Corporativas PPT 1Documento70 páginasFinanzas Corporativas PPT 1Nataly Mendoza DiazAún no hay calificaciones
- 0381a-Mpes-C-2022 Mar Andino Perú Sac 30 KG X 0,005 KGDocumento3 páginas0381a-Mpes-C-2022 Mar Andino Perú Sac 30 KG X 0,005 KGJose Luis CamachoAún no hay calificaciones
- 4ta LecturaDocumento32 páginas4ta LecturaWendy de Ángeles Oliveros CarmonaAún no hay calificaciones
- Nic 37Documento34 páginasNic 37Juan Antonio Donoso vergaraAún no hay calificaciones
- Riesgo y RendimientoDocumento10 páginasRiesgo y RendimientoCarlos SantosAún no hay calificaciones
- Tecnicas de Planificacion de RiesgosDocumento110 páginasTecnicas de Planificacion de RiesgoscarolAún no hay calificaciones
- Caracteristicas Estadicas y DinamicasDocumento32 páginasCaracteristicas Estadicas y DinamicasJose Camilo Eraso GuerreroAún no hay calificaciones
- Práctica (Unidad 3), Contabilidad GubernamentalDocumento6 páginasPráctica (Unidad 3), Contabilidad GubernamentalMiguelinaAún no hay calificaciones
- Programa de Ice, Revisado 2015Documento5 páginasPrograma de Ice, Revisado 2015Ernesto RamirezAún no hay calificaciones
- LAB-2020-1 Mediciones ExperimentalDocumento4 páginasLAB-2020-1 Mediciones ExperimentalFLOR CARIZA URACCAHUA CABRERAAún no hay calificaciones
- Informe 1 FisicaDocumento7 páginasInforme 1 FisicaJOEL DAVID HERNANDEZ SARMIENTOAún no hay calificaciones
- Gestión Cadena de Abastecimiento - Logistica Con Indicadores Bajo Incertidumbre, Caso Aplicado Sector Panificador PalmiraDocumento14 páginasGestión Cadena de Abastecimiento - Logistica Con Indicadores Bajo Incertidumbre, Caso Aplicado Sector Panificador PalmiraARMANDOAún no hay calificaciones
- El Sistema GYRO Un Enfoque Recomendado para Una DocumentaciónDocumento9 páginasEl Sistema GYRO Un Enfoque Recomendado para Una Documentaciónezain rodriguezAún no hay calificaciones
- Informe Laboratorio 8 Fisica Imáquinas SimplesDocumento20 páginasInforme Laboratorio 8 Fisica Imáquinas SimplesManuel Jose Arango MoncadaAún no hay calificaciones
- MC-DT-P-002 Procedimiento Estimacion IncertidumbreDocumento8 páginasMC-DT-P-002 Procedimiento Estimacion IncertidumbreDireccion TecnicaAún no hay calificaciones
- Práctica 1 - Mediciones e IncertidumbreDocumento9 páginasPráctica 1 - Mediciones e IncertidumbreCarlos BernedoAún no hay calificaciones
- Sistema Automatizado de Calibración de Densímetros de Inmersión para El Organismo Nacional de Metrología Del Instituto Nacional de Tecnología, Normalización y Metrología (Intn)Documento173 páginasSistema Automatizado de Calibración de Densímetros de Inmersión para El Organismo Nacional de Metrología Del Instituto Nacional de Tecnología, Normalización y Metrología (Intn)Gustavo Adelio Riveiro GonzalezAún no hay calificaciones
- Mejoramiento de la calidad. Un enfoque a serviciosDe EverandMejoramiento de la calidad. Un enfoque a serviciosCalificación: 4.5 de 5 estrellas4.5/5 (7)
- Marketing de contenidos. Guía prácticaDe EverandMarketing de contenidos. Guía prácticaCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Prótesis dental artesanal de calidad: Juntos hacía el éxito: unimos disciplinasDe EverandPrótesis dental artesanal de calidad: Juntos hacía el éxito: unimos disciplinasCalificación: 3 de 5 estrellas3/5 (1)
- Calidad y servicio. Concepto y herramientasDe EverandCalidad y servicio. Concepto y herramientasAún no hay calificaciones
- Fundamentos de producción y gestión de proyectos audiovisualesDe EverandFundamentos de producción y gestión de proyectos audiovisualesCalificación: 5 de 5 estrellas5/5 (2)
- Manual para manipuladores de alimentos: InstructorDe EverandManual para manipuladores de alimentos: InstructorCalificación: 4.5 de 5 estrellas4.5/5 (4)
- Calidad y servicio: Conceptos y herramientasDe EverandCalidad y servicio: Conceptos y herramientasCalificación: 5 de 5 estrellas5/5 (2)
- Gestión de la calidad ISO 9001/2015 en hosteleriaDe EverandGestión de la calidad ISO 9001/2015 en hosteleriaCalificación: 5 de 5 estrellas5/5 (1)
- UF0285 - Tratamiento de residuos urbanos o municipales.De EverandUF0285 - Tratamiento de residuos urbanos o municipales.Aún no hay calificaciones
- UF1941 - Elaboración de inventarios de focos contaminantesDe EverandUF1941 - Elaboración de inventarios de focos contaminantesCalificación: 5 de 5 estrellas5/5 (1)
- El concepto de calidad en la organización y su sentido estratégicoDe EverandEl concepto de calidad en la organización y su sentido estratégicoCalificación: 4.5 de 5 estrellas4.5/5 (6)
- Gestión Administrativa y Comercial en RestauraciónDe EverandGestión Administrativa y Comercial en RestauraciónAún no hay calificaciones
- Basurascopio: Una exploración al mundo de la basuraDe EverandBasurascopio: Una exploración al mundo de la basuraCalificación: 5 de 5 estrellas5/5 (3)
- El método Seis Sigma: Mejore los resultados de su negocioDe EverandEl método Seis Sigma: Mejore los resultados de su negocioCalificación: 4 de 5 estrellas4/5 (24)
- UF2119 - Planificación de la investigación de mercadosDe EverandUF2119 - Planificación de la investigación de mercadosAún no hay calificaciones
- UF1666 - Depuración de aguas residualesDe EverandUF1666 - Depuración de aguas residualesAún no hay calificaciones
- UF0284 - Recogida y transporte de residuos urbanos o municipalesDe EverandUF0284 - Recogida y transporte de residuos urbanos o municipalesAún no hay calificaciones
- Gestión de la calidad ISO 9001/2015 en comercioDe EverandGestión de la calidad ISO 9001/2015 en comercioAún no hay calificaciones
- La Gestión de los Residuos Sólidos Urbanos en Brasil:: descripción general, conceptos, aplicaciones y perspectivasDe EverandLa Gestión de los Residuos Sólidos Urbanos en Brasil:: descripción general, conceptos, aplicaciones y perspectivasCalificación: 1 de 5 estrellas1/5 (1)
- Conceptos Básicos De Scrum: Desarrollo De Software Agile Y Manejo De Proyectos AgileDe EverandConceptos Básicos De Scrum: Desarrollo De Software Agile Y Manejo De Proyectos AgileCalificación: 5 de 5 estrellas5/5 (6)
- Historia del Movimiento Ambiental en Puerto RicoDe EverandHistoria del Movimiento Ambiental en Puerto RicoCalificación: 4.5 de 5 estrellas4.5/5 (2)
- Estrategias de muestreo: Diseño de encuestas y estimación de parámetrosDe EverandEstrategias de muestreo: Diseño de encuestas y estimación de parámetrosCalificación: 5 de 5 estrellas5/5 (1)
- UF1671 - Mantenimiento del entorno de plantas de tratamiento de agua y plantas depuradorasDe EverandUF1671 - Mantenimiento del entorno de plantas de tratamiento de agua y plantas depuradorasCalificación: 5 de 5 estrellas5/5 (1)
- Modelamiento logístico para la producción sostenible de biocombustiblesDe EverandModelamiento logístico para la producción sostenible de biocombustiblesCalificación: 4 de 5 estrellas4/5 (1)
- Manual para manipuladores de alimentos: AlumnoDe EverandManual para manipuladores de alimentos: AlumnoAún no hay calificaciones
- Las 3 R para disminuir la contaminación ambientalDe EverandLas 3 R para disminuir la contaminación ambientalCalificación: 5 de 5 estrellas5/5 (1)
- Diseño institucional de las entidades de fiscalización superior de América LatinaDe EverandDiseño institucional de las entidades de fiscalización superior de América LatinaCalificación: 5 de 5 estrellas5/5 (1)
- Transversalidad de la política de la calidad del aire en MéxicoDe EverandTransversalidad de la política de la calidad del aire en MéxicoAún no hay calificaciones
- Ingeniería ambiental: Manejo de ecosistemas, concepción de políticas públicas y reciclaje de materialesDe EverandIngeniería ambiental: Manejo de ecosistemas, concepción de políticas públicas y reciclaje de materialesCalificación: 4 de 5 estrellas4/5 (1)