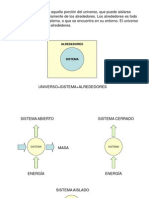
También podría gustarte
- 1585698262.T - 1 - Combinacion Del Primer y Seg PrincipioDocumento38 páginas1585698262.T - 1 - Combinacion Del Primer y Seg PrincipioPedro InsaurraldeAún no hay calificaciones
- Cuestionario de Primeley en Sis Temas CerradosDocumento8 páginasCuestionario de Primeley en Sis Temas CerradosKarina Fernanda Torres RosalesAún no hay calificaciones
- Semana 2 2012Documento8 páginasSemana 2 2012quimicocad9891Aún no hay calificaciones
- Parte 22. TERCER PRINCIPIO DE TERMODINÁMICADocumento17 páginasParte 22. TERCER PRINCIPIO DE TERMODINÁMICAErik Ariel LauraAún no hay calificaciones
- Clase2 PDFDocumento7 páginasClase2 PDFMATIAS GABRIEL RICCIAún no hay calificaciones
- Soluciones Examenes2223Documento38 páginasSoluciones Examenes2223Luis SuarezAún no hay calificaciones
- Apuntes TermoDocumento7 páginasApuntes TermoLinda Jaqueline Narvaez OchoaAún no hay calificaciones
- Clase 4Documento8 páginasClase 4botametunosegaAún no hay calificaciones
- Módulo Viii - Segundo Principio de La TermodinámicaDocumento28 páginasMódulo Viii - Segundo Principio de La TermodinámicaIsaac MuñozAún no hay calificaciones
- Unidad 2. Termodinámica IIDocumento57 páginasUnidad 2. Termodinámica IIJorge GallardoAún no hay calificaciones
- Examen de Termodinamica ETSII UPV ValenciaDocumento7 páginasExamen de Termodinamica ETSII UPV ValenciaAaron Molina GalanAún no hay calificaciones
- Procesos Fisicos de GasesDocumento40 páginasProcesos Fisicos de GasesPaulina SantanaAún no hay calificaciones
- INTRODUCCIÓNDocumento22 páginasINTRODUCCIÓNISAI HUACHOS BENDEZUAún no hay calificaciones
- 7 Termot Exergia 2010 11Documento32 páginas7 Termot Exergia 2010 11Jorge Perez GarciaAún no hay calificaciones
- Transcripcion 13Documento25 páginasTranscripcion 13Luis MorilloAún no hay calificaciones
- Control e Instrumentación de Procesos Químicos:) 2 T (Bu VT DT DV VDocumento67 páginasControl e Instrumentación de Procesos Químicos:) 2 T (Bu VT DT DV VRenzo PinedoAún no hay calificaciones
- Semana N°3 - Primera Ley de La TermodinámicaDocumento20 páginasSemana N°3 - Primera Ley de La TermodinámicaLesly PeredaAún no hay calificaciones
- Relaciones de MaxwellDocumento19 páginasRelaciones de MaxwellSamuel Parra RestrepoAún no hay calificaciones
- Principios de TermodinamicaDocumento5 páginasPrincipios de TermodinamicaLuis David Brito Saldivar100% (1)
- Termo ReversibilidadDocumento16 páginasTermo ReversibilidadManuel Alejandro PeñafielAún no hay calificaciones
- Diseño de Reactores No Isotérmicos en Estado EstacionarioDocumento8 páginasDiseño de Reactores No Isotérmicos en Estado EstacionarioJulieth JiménezAún no hay calificaciones
- Resumen Primera Ley de La TermodinámicaDocumento3 páginasResumen Primera Ley de La TermodinámicaAndree AlmeidaAún no hay calificaciones
- Fisica Solemne 2Documento5 páginasFisica Solemne 2Diego Barra AlvarezAún no hay calificaciones
- Termodinamica UdeMM - 7Documento20 páginasTermodinamica UdeMM - 7Martin Javier RadogowskiAún no hay calificaciones
- Analisis Solido Semi InfinitoDocumento13 páginasAnalisis Solido Semi InfinitoGRANRICKY100% (1)
- Bolilla 6Documento13 páginasBolilla 6Facundo EscobarAún no hay calificaciones
- ENTROPIADocumento5 páginasENTROPIAJhonatan Meza AparicioAún no hay calificaciones
- Equilibrio-MaterialDocumento67 páginasEquilibrio-MaterialIVETAún no hay calificaciones
- Equilibrio Material PDFDocumento10 páginasEquilibrio Material PDFPercyD.RojasAún no hay calificaciones
- Formula RioDocumento17 páginasFormula RioCristian Lema BañaAún no hay calificaciones
- ExergiaDocumento12 páginasExergiaCursos Facultad de IngenieríaAún no hay calificaciones
- Unidad Iv-1 Ley Termo - 1Documento33 páginasUnidad Iv-1 Ley Termo - 1Oscar RodriguezAún no hay calificaciones
- Unidad 2. Diseño de Reactores Isotérmicos y No IsotérmicosDocumento7 páginasUnidad 2. Diseño de Reactores Isotérmicos y No IsotérmicosEsteban Fernando Carrilo VazquezAún no hay calificaciones
- CAPÍTULO TRABAJO Y COLOR - TermodinamicaDocumento11 páginasCAPÍTULO TRABAJO Y COLOR - Termodinamicaapuatau94620% (2)
- Ecuación de ClapeyronDocumento1 páginaEcuación de Clapeyrongabriel riveroAún no hay calificaciones
- L001 PDFDocumento4 páginasL001 PDFGunnar GutierrezAún no hay calificaciones
- Tarea y Practica 10 Transformacion AdiabaticaDocumento10 páginasTarea y Practica 10 Transformacion AdiabaticaDelaia Nicole Ulloa AriasAún no hay calificaciones
- Formulario de Termodinámica.Documento5 páginasFormulario de Termodinámica.AbrahanguerraAún no hay calificaciones
- Segunda, Tercera Ley y Energía LibreDocumento18 páginasSegunda, Tercera Ley y Energía LibreCESAR ANTONIO DIAZ CISNEROSAún no hay calificaciones
- Primera Ley de TermodinamicaDocumento71 páginasPrimera Ley de TermodinamicaAndreaAún no hay calificaciones
- Guia Numero 5 - 2022 - Entropia y ExergiaDocumento10 páginasGuia Numero 5 - 2022 - Entropia y Exergiapedrito mmmaaAún no hay calificaciones
- Condtransitoria2 PDFDocumento53 páginasCondtransitoria2 PDFJulio Andres HerreraAún no hay calificaciones
- Primera Ley Problemas Resueltos TermodinamicaDocumento27 páginasPrimera Ley Problemas Resueltos TermodinamicaJuvenal GalarceAún no hay calificaciones
- Portafolio 26 y 29Documento13 páginasPortafolio 26 y 29MoyCoral100% (3)
- Práctica #3Documento29 páginasPráctica #3SofiaAguirreAún no hay calificaciones
- Energía LibreDocumento35 páginasEnergía Librechio_garcia_2Aún no hay calificaciones
- Problemas Del PathriaDocumento13 páginasProblemas Del PathriaMontse CrespoAún no hay calificaciones
- Equilibrio QuimicoDocumento26 páginasEquilibrio QuimicoPabloEzequielRamosAún no hay calificaciones
- T 2 Primer Principio de La Termodinámica (Teoría)Documento29 páginasT 2 Primer Principio de La Termodinámica (Teoría)Tatiana ZorrillaAún no hay calificaciones
- VI. - Conducción de Calor Transitoria en Sólidos SemiinfinitosDocumento13 páginasVI. - Conducción de Calor Transitoria en Sólidos SemiinfinitosFrancisco González De Brito100% (3)
- 13 Analisis Exergético 2 PDFDocumento20 páginas13 Analisis Exergético 2 PDFosmar yordyAún no hay calificaciones
- Semana 9Documento3 páginasSemana 9Marcio BautistaAún no hay calificaciones
- Unidad IVDocumento6 páginasUnidad IVcanaima linuxAún no hay calificaciones
- Tema 2 - 2016 QuifiDocumento45 páginasTema 2 - 2016 QuifiisaAún no hay calificaciones
- Expansión de Joule-ThomsonDocumento7 páginasExpansión de Joule-Thomsonleslie gianella dominguez chavezAún no hay calificaciones
- Problemas resueltos de Hidráulica de CanalesDe EverandProblemas resueltos de Hidráulica de CanalesCalificación: 4.5 de 5 estrellas4.5/5 (7)
- Ingeniería química. Soluciones a los problemas del tomo IDe EverandIngeniería química. Soluciones a los problemas del tomo IAún no hay calificaciones
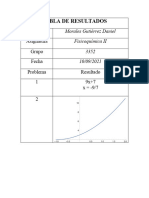
- Tabla de ResultadosDocumento2 páginasTabla de ResultadosLOBOBLOXYT EL CRAK2000Aún no hay calificaciones
- Cuaderno de Separación Mécanica y MezcladoDocumento58 páginasCuaderno de Separación Mécanica y MezcladoLOBOBLOXYT EL CRAK2000Aún no hay calificaciones
- Apuntes Unidad 1Documento138 páginasApuntes Unidad 1LOBOBLOXYT EL CRAK2000Aún no hay calificaciones
- Apuntes Unidad 2Documento105 páginasApuntes Unidad 2LOBOBLOXYT EL CRAK2000Aún no hay calificaciones
- Membrana Celular y TransporteDocumento8 páginasMembrana Celular y TransporteMili ParodiAún no hay calificaciones
- Easy UPS - BV500I MS 1Documento3 páginasEasy UPS - BV500I MS 1WILMER GUTIERREZAún no hay calificaciones
- Diferencias Entre Inversores de Onda Pura y de Onda ModificadaDocumento2 páginasDiferencias Entre Inversores de Onda Pura y de Onda ModificadaM a r t i nAún no hay calificaciones
- Catalogo General Legrand 2021-2022Documento868 páginasCatalogo General Legrand 2021-2022Roberto GutierrezAún no hay calificaciones
- Compact NS - 630 A - 33466Documento3 páginasCompact NS - 630 A - 33466Jhonatan Fabian Cely AparicioAún no hay calificaciones
- Calculo de Espesores de Pared Segun Normas B31Documento33 páginasCalculo de Espesores de Pared Segun Normas B31Edinson Rolando Rodriguez MondragonAún no hay calificaciones
- Contexto Histórico de La Industria 4.0Documento6 páginasContexto Histórico de La Industria 4.0Fátima CarrilloAún no hay calificaciones
- Informe Final Tambobamba OkDocumento42 páginasInforme Final Tambobamba OkCiro Avalos DuranAún no hay calificaciones
- Observaciones de La Cotizacion de La TurbinaDocumento7 páginasObservaciones de La Cotizacion de La TurbinaLevi AckermanAún no hay calificaciones
- Laboratorio de Automatización Industrial NDocumento3 páginasLaboratorio de Automatización Industrial NJhonny Edgar Mamani QuispeAún no hay calificaciones
- 03 ES Schematy Podlaczen Instalacji ALEX Optima ExpertDocumento5 páginas03 ES Schematy Podlaczen Instalacji ALEX Optima ExpertNorberto CaponigroAún no hay calificaciones
- Propiedades de Una Sustancia PuraDocumento14 páginasPropiedades de Una Sustancia PuraLuisAún no hay calificaciones
- Experiencia en Uso de Aisladores PolimericosDocumento6 páginasExperiencia en Uso de Aisladores PolimericosjorgemanuicoAún no hay calificaciones
- Formulario CombustionDocumento1 páginaFormulario CombustionEmmanuell KeithAún no hay calificaciones
- Estudio de Prefactibilidad Tecnica y Economica de La Produccion de Hidrógeno Mediante Electrolisis para La Entidad GnaDocumento5 páginasEstudio de Prefactibilidad Tecnica y Economica de La Produccion de Hidrógeno Mediante Electrolisis para La Entidad GnaJean Piere SotoAún no hay calificaciones
- Tarea 2 - 2 AxcellPerdidasDelMotorDocumento3 páginasTarea 2 - 2 AxcellPerdidasDelMotorAldo David Silva MartinezAún no hay calificaciones
- Propuesta para La Integración de Criterios Sostenibles en Los Proyectos de Ingeniería Civil - Un Caso PrácticoDocumento10 páginasPropuesta para La Integración de Criterios Sostenibles en Los Proyectos de Ingeniería Civil - Un Caso PrácticoEnrique Gutierrez LopezAún no hay calificaciones
- Convertidores Ca-Ca DirectosDocumento10 páginasConvertidores Ca-Ca DirectosERIK HERNANDEZ HERNANDEZAún no hay calificaciones
- Inyectores Bosch Partes de RecambioDocumento3 páginasInyectores Bosch Partes de RecambioJosue SilveiraAún no hay calificaciones
- RECOCIDODocumento7 páginasRECOCIDOLuisa LopezAún no hay calificaciones
- Preguntero 2do ParcialDocumento9 páginasPreguntero 2do ParcialGime SaddlerAún no hay calificaciones
- Proyecto de Investigación - Juan Carlos Suarez ZetaDocumento26 páginasProyecto de Investigación - Juan Carlos Suarez Zetaluis SuarezAún no hay calificaciones
- Clasificación de La MateriaDocumento8 páginasClasificación de La MateriaBrayan BarrientosAún no hay calificaciones
- Estudio de Casos de Empresas Colombianas Parte 2.Documento16 páginasEstudio de Casos de Empresas Colombianas Parte 2.Jhon Edwin Lozano AlvarezAún no hay calificaciones
- Alambre Cable CordonDocumento3 páginasAlambre Cable CordonhebertAún no hay calificaciones
- PXL-GDSSSTPA-PCS-06 Bloqueo de Energia y Materiales PeligrososDocumento41 páginasPXL-GDSSSTPA-PCS-06 Bloqueo de Energia y Materiales PeligrososJuan Andrés González Montero100% (1)
- Estados de Agregación y FénomenosDocumento5 páginasEstados de Agregación y FénomenosSantiago RubioAún no hay calificaciones
- Motores y Alternador SíncronoDocumento13 páginasMotores y Alternador SíncronoIgnacio ArandaAún no hay calificaciones
- Anual San Marcos Fisica Semana21Documento12 páginasAnual San Marcos Fisica Semana21Katarine LunaAún no hay calificaciones
- 11 Sturm CFE Ejemplos de FGVDocumento14 páginas11 Sturm CFE Ejemplos de FGVIsra LopezAún no hay calificaciones