También podría gustarte
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Regulacion Agua y Electrolitos - Casos ClinicosDocumento12 páginasRegulacion Agua y Electrolitos - Casos ClinicosCAROL NATALIA PIÑEROS JIMENEZ0% (1)
- Disnatremias. Pablo CastañoDocumento25 páginasDisnatremias. Pablo CastañoJudyAún no hay calificaciones
- HiponatremiaDocumento22 páginasHiponatremiaivantico38Aún no hay calificaciones
- Formas Sencillas De Detener El Reflujo Ácido: Lo que nadie te cuenta acerca de cómo luchar con esta enfermedadDe EverandFormas Sencillas De Detener El Reflujo Ácido: Lo que nadie te cuenta acerca de cómo luchar con esta enfermedadCalificación: 5 de 5 estrellas5/5 (2)
- SIADHDocumento38 páginasSIADHMadeleine GilAún no hay calificaciones
- Soluciones para la Diabetes y la Hipoglucemia (Traducido): Cómo prevenirla y deshacerse de ella de forma natural, sin medicamentos pero adoptando un estilo de vida saludableDe EverandSoluciones para la Diabetes y la Hipoglucemia (Traducido): Cómo prevenirla y deshacerse de ella de forma natural, sin medicamentos pero adoptando un estilo de vida saludableAún no hay calificaciones
- Autonomo 1Documento5 páginasAutonomo 1Valeria BueleAún no hay calificaciones
- Enfoque Diagnóstico de La Hipocalcemia: IntroducciónDocumento16 páginasEnfoque Diagnóstico de La Hipocalcemia: IntroducciónMateo GranjaAún no hay calificaciones
- Interpretación de análisis clínicos en perros y gatosDe EverandInterpretación de análisis clínicos en perros y gatosCalificación: 5 de 5 estrellas5/5 (2)
- Ex Lab Relacion Bun Crea Baja P 2Documento5 páginasEx Lab Relacion Bun Crea Baja P 2Marco PulidoAún no hay calificaciones
- S1. Caso Clínico 55Documento7 páginasS1. Caso Clínico 55tere ariasAún no hay calificaciones
- Electrolitos y Gases ArterialesDocumento37 páginasElectrolitos y Gases ArterialesRomulo ChuquillanquiAún no hay calificaciones
- RAE CCL3 HiponatremiaDocumento8 páginasRAE CCL3 Hiponatremiacamila rodriguezAún no hay calificaciones
- Resumen CL 3Documento8 páginasResumen CL 3Laura Ospina SantosAún no hay calificaciones
- Trastornos de Metabolismo de SodioDocumento7 páginasTrastornos de Metabolismo de SodioSarahi UlloaAún no hay calificaciones
- Trastornos Del PotasioDocumento9 páginasTrastornos Del PotasioSofi PetroAún no hay calificaciones
- Enfermedad de AddisonDocumento18 páginasEnfermedad de AddisonMalena WeinsingerAún no hay calificaciones
- Nefrologia Grupo 2Documento36 páginasNefrologia Grupo 2César ValdiviaAún no hay calificaciones
- Casos Mixtos Alcalosis y AcidosisDocumento12 páginasCasos Mixtos Alcalosis y AcidosisMaritza LPAún no hay calificaciones
- Trastornos Del Potasio: Puntos de Buena Práctica Clínica AutorDocumento17 páginasTrastornos Del Potasio: Puntos de Buena Práctica Clínica AutorAndrés MontalvoAún no hay calificaciones
- Síndrome Cerebral Perdedor de SalDocumento5 páginasSíndrome Cerebral Perdedor de Salarnaldops333Aún no hay calificaciones
- Seminario 5 - Fisiología Endocrina 1Documento9 páginasSeminario 5 - Fisiología Endocrina 1Fernanda Elizabeth MolinaAún no hay calificaciones
- Desgloses NF PDFDocumento14 páginasDesgloses NF PDFalma0pineda100% (1)
- Hiponatremia Patofisiología PDFDocumento22 páginasHiponatremia Patofisiología PDFDiana SofíaAún no hay calificaciones
- Acidosis MetabolicaDocumento5 páginasAcidosis MetabolicaFatima AlvaradoAún no hay calificaciones
- Preguntas de Las ClasesDocumento4 páginasPreguntas de Las Clasesrrrm1968Aún no hay calificaciones
- Caso Clino 8 FisiopatoDocumento7 páginasCaso Clino 8 FisiopatoMaria VelaAún no hay calificaciones
- Informe 3 Bioquimíca PrácticaDocumento10 páginasInforme 3 Bioquimíca PrácticaEmery Sofía C. D. BustamanteAún no hay calificaciones
- Electrolitos Sanguíneos Y Análisis de Gases Arteriales-MONOGRAFIADocumento47 páginasElectrolitos Sanguíneos Y Análisis de Gases Arteriales-MONOGRAFIAalan_05068350% (4)
- Diabetes Manejo Crisis Hiperglicemicas Ada - NewDocumento9 páginasDiabetes Manejo Crisis Hiperglicemicas Ada - NewIttan GloverAún no hay calificaciones
- Nefrología Examen 2 CDocumento5 páginasNefrología Examen 2 CSteph OsAún no hay calificaciones
- Diagnóstico y Manejo de La Crisis HiperglucémicaDocumento9 páginasDiagnóstico y Manejo de La Crisis HiperglucémicaFrancisco MontañoAún no hay calificaciones
- ComentarioDocumento14 páginasComentarioGonzalo LaricoAún no hay calificaciones
- Caso Clínico N°6Documento19 páginasCaso Clínico N°6DIANA LUCIA CHIMOY VILLANUEVAAún no hay calificaciones
- Endocrinologia Examen ENAM - PLUS MedicaDocumento8 páginasEndocrinologia Examen ENAM - PLUS MedicaMaria Gracia Pereda GinocchioAún no hay calificaciones
- Objetivos de La BioquímicaDocumento44 páginasObjetivos de La BioquímicaDitza MarianAún no hay calificaciones
- Flujo UrinarioDocumento4 páginasFlujo UrinariomaxAún no hay calificaciones
- Hiponatremia e Hipernatremia José Peña 2012 PDFDocumento16 páginasHiponatremia e Hipernatremia José Peña 2012 PDFCarola Muñoz Arratia.Aún no hay calificaciones
- Cetoacidosis OsvyDocumento33 páginasCetoacidosis OsvyLeidy Mujica RojasAún no hay calificaciones
- Práctica 10. Cuantificación de Ácido ÚricoDocumento6 páginasPráctica 10. Cuantificación de Ácido ÚricoJesús SalazarAún no hay calificaciones
- Examen Medicina InternaDocumento75 páginasExamen Medicina InternaAngelica BolañosAún no hay calificaciones
- Saladin 6a Caso c14 Fisiologia AcidobasicaDocumento4 páginasSaladin 6a Caso c14 Fisiologia AcidobasicaMAXI PLUS 1Aún no hay calificaciones
- CASOS CLINICOS III ParcialDocumento9 páginasCASOS CLINICOS III Parcialanon_927936041Aún no hay calificaciones
- Electrolitos Sanguineos Y Analisis de Gases Arteriales MONOGRAFIADocumento47 páginasElectrolitos Sanguineos Y Analisis de Gases Arteriales MONOGRAFIACarlos RojasAún no hay calificaciones
- Informe de Grupo 4 Trastornos ElectrolíticosDocumento23 páginasInforme de Grupo 4 Trastornos ElectrolíticosAV AVAún no hay calificaciones
- FISIOPATOLOGIA2Documento61 páginasFISIOPATOLOGIA2medaly de la cruzAún no hay calificaciones
- Nefrologia Civer 2020Documento270 páginasNefrologia Civer 2020jobperuAún no hay calificaciones
- Balance HidricoDocumento13 páginasBalance HidricoMarjorie Natalia Ururi OréAún no hay calificaciones
- ALBUM Quimica SanguineaDocumento24 páginasALBUM Quimica SanguineaJovas R. Moreno De RechyAún no hay calificaciones
- Manejo Crisis HiperglicemicasDocumento7 páginasManejo Crisis HiperglicemicasKaren Campos ManjaresAún no hay calificaciones
- Desequilibrio HidroelectrolíticoDocumento20 páginasDesequilibrio Hidroelectrolíticoesperanza.guzman.umeAún no hay calificaciones
- Banco Preguntas - Onco, Reumato, EndocrinoDocumento5 páginasBanco Preguntas - Onco, Reumato, EndocrinoRaquel Karina HuamánAún no hay calificaciones
- Clase Práctica. ContestadasDocumento4 páginasClase Práctica. ContestadasMilena LoaisigaAún no hay calificaciones
- Electrolitos SodioDocumento14 páginasElectrolitos SodioEhyleen Romero VillarrealAún no hay calificaciones
- Electrolitos Sanguineos Y Analisis de Gases Arteriales MONOGRAFIADocumento48 páginasElectrolitos Sanguineos Y Analisis de Gases Arteriales MONOGRAFIAJohanaAlbeaAún no hay calificaciones
- Casos Clinicos de Ac-Bas y HidroDocumento54 páginasCasos Clinicos de Ac-Bas y HidroBarbara Daniela Gonzalez Espinoza0% (1)
- 1A. - Caso Clinico Análisis Espermático (Maquetado)Documento5 páginas1A. - Caso Clinico Análisis Espermático (Maquetado)tere ariasAún no hay calificaciones
- Tema-56-Rev. 10-22 JuliaDocumento16 páginasTema-56-Rev. 10-22 Juliatere ariasAún no hay calificaciones
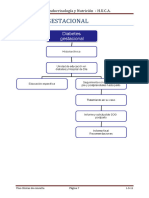
- Diabetes Gestacional: S. de Endocrinología y Nutrición - H.U.C.ADocumento1 páginaDiabetes Gestacional: S. de Endocrinología y Nutrición - H.U.C.Atere ariasAún no hay calificaciones
- Determinación de Proteínas UD5Documento46 páginasDeterminación de Proteínas UD5tere ariasAún no hay calificaciones
- Tema-57-Rev10-22 JuliaDocumento24 páginasTema-57-Rev10-22 Juliatere ariasAún no hay calificaciones
- S1. Caso Clínico 69Documento7 páginasS1. Caso Clínico 69tere ariasAún no hay calificaciones
- Caso Clínico 73Documento8 páginasCaso Clínico 73tere ariasAún no hay calificaciones
- S1. Caso Clínico 62Documento4 páginasS1. Caso Clínico 62tere ariasAún no hay calificaciones
- S1. Caso Clínico 55Documento7 páginasS1. Caso Clínico 55tere ariasAún no hay calificaciones
- S1. Caso Clínico 52Documento8 páginasS1. Caso Clínico 52tere ariasAún no hay calificaciones
- Estadistica Cancer Tiroides HRGGB 2010-2014Documento9 páginasEstadistica Cancer Tiroides HRGGB 2010-2014Cristopher PalmaAún no hay calificaciones
- Histología de Organos GenitalesDocumento63 páginasHistología de Organos GenitalesEmanuel PatricioAún no hay calificaciones
- Sistema EndocrinoDocumento1 páginaSistema EndocrinoBRISA LIZETT YUPIT ALVAREZAún no hay calificaciones
- HipertiroidismoDocumento11 páginasHipertiroidismoEric Gomez CortesAún no hay calificaciones
- Act 9 B 2 Sistema EndocrinoDocumento1 páginaAct 9 B 2 Sistema EndocrinoPablo Andrik Martín SalvadorAún no hay calificaciones
- Gónadas MasculinasDocumento7 páginasGónadas Masculinasjoshua0% (1)
- RM 20 F2 - Endocrinología 1 - OnlineDocumento28 páginasRM 20 F2 - Endocrinología 1 - OnlineAlex Vigo NovoaAún no hay calificaciones
- Examen PDFDocumento7 páginasExamen PDFLuis CoteAún no hay calificaciones
- TIROIDECTOMIADocumento28 páginasTIROIDECTOMIAPatricia Maria Lea AlmanzaAún no hay calificaciones
- C TiroidesDocumento88 páginasC TiroidesStefhany ContrerasAún no hay calificaciones
- Tema 46 - Carcinoma Diferenciado TiroidesDocumento41 páginasTema 46 - Carcinoma Diferenciado Tiroidesapi-3705495100% (2)
- TerapiaPostCiclo - PDF HSPDocumento42 páginasTerapiaPostCiclo - PDF HSPErick Bertha Miranda CaicedoAún no hay calificaciones
- Tiroides ActualizadoDocumento54 páginasTiroides ActualizadoAdriana CastañedaAún no hay calificaciones
- Examenes de La Laboratorio 28 Febrero 2016 PDFDocumento1 páginaExamenes de La Laboratorio 28 Febrero 2016 PDFYuri Osorio MedinaAún no hay calificaciones
- AMENORREADocumento15 páginasAMENORREAMishelV06Aún no hay calificaciones
- Guía - Sistema Endocrino y Su Relacion Con La HomeostasisDocumento19 páginasGuía - Sistema Endocrino y Su Relacion Con La HomeostasisAlexander Donado MolinaresAún no hay calificaciones
- Adolescencia y PubertadDocumento37 páginasAdolescencia y PubertadDIEGO ALEXANDER HERRADA URRIAGOAún no hay calificaciones
- Glandulas Endocrinas PROYECTO INVESTIGACION, - Eduardo Cruz.Documento69 páginasGlandulas Endocrinas PROYECTO INVESTIGACION, - Eduardo Cruz.Alejo De LeonAún no hay calificaciones
- Síndrome de CushingDocumento10 páginasSíndrome de CushingCLARA FERNANDA REYES NEGRETEAún no hay calificaciones
- Desarollo y Comportamiento Sexual PDFDocumento16 páginasDesarollo y Comportamiento Sexual PDFDavid SandovalAún no hay calificaciones
- Granados Gutierrez Sandra PatriciaDocumento1 páginaGranados Gutierrez Sandra PatriciaDaniel MrtinezAún no hay calificaciones
- Endocrinologiaexamen FinalDocumento262 páginasEndocrinologiaexamen FinalBrandon RyanAún no hay calificaciones
- Emilio Diaz VillegasDocumento2 páginasEmilio Diaz VillegasLuis Javier Martinez EscuderoAún no hay calificaciones
- GS-D.290.00 Protocolo de HipotiroidismoDocumento8 páginasGS-D.290.00 Protocolo de HipotiroidismoDIVY LUZ HERNANDEZAún no hay calificaciones
- SEMIOLOGÍA TIROIDEA 1noDocumento45 páginasSEMIOLOGÍA TIROIDEA 1noEver Andres Zegarra CandiottiAún no hay calificaciones
- Ejemplo AuditoriaDocumento165 páginasEjemplo AuditoriaErika Lissette MEDINA TRIANAAún no hay calificaciones
- Hormonas y Actividad FísicaDocumento6 páginasHormonas y Actividad FísicaZulma Andrea Pescador VillegasAún no hay calificaciones
- Bioquímica II Quinta ClaseDocumento33 páginasBioquímica II Quinta ClasePercy J DiazAún no hay calificaciones
- 6° Grado - Actividad Del Dia 03 de JunioDocumento39 páginas6° Grado - Actividad Del Dia 03 de Junioana de la cruzAún no hay calificaciones
- Informe Sistema EndocrinoDocumento15 páginasInforme Sistema EndocrinoericaAún no hay calificaciones
- Muchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)De EverandMuchas Vidas, Muchos Sabios (Many Lives, Many Masters): (Many Lives, Many Masters)Calificación: 4 de 5 estrellas4/5 (475)
- El concepto Mulligan de terapia manual (Color)De EverandEl concepto Mulligan de terapia manual (Color)Calificación: 5 de 5 estrellas5/5 (3)
- Ondas de choque extracorpóreas radiales: Aplicación en patologías músculo esqueléticasDe EverandOndas de choque extracorpóreas radiales: Aplicación en patologías músculo esqueléticasCalificación: 5 de 5 estrellas5/5 (3)
- El código de la obesidad: Descifrando los secretos de la pérdida de pesoDe EverandEl código de la obesidad: Descifrando los secretos de la pérdida de pesoCalificación: 4.5 de 5 estrellas4.5/5 (51)
- Masaje de los tejidos profundos: Guía visual de las técnicasDe EverandMasaje de los tejidos profundos: Guía visual de las técnicasCalificación: 3 de 5 estrellas3/5 (6)
- La metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceDe EverandLa metamedicina. Cada síntoma es un mensaje: La curación a tu alcanceCalificación: 5 de 5 estrellas5/5 (8)
- La medicina biorreguladora: Un enfoque holístico e innovador de la autocuraciónDe EverandLa medicina biorreguladora: Un enfoque holístico e innovador de la autocuraciónCalificación: 3.5 de 5 estrellas3.5/5 (2)
- GuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisDe EverandGuíaBurros Análisis clínicos: Todo lo que necesitas saber para entender tus análisisCalificación: 4 de 5 estrellas4/5 (9)
- Vivir con endometriosis: Una guía para recuperar el bienestarDe EverandVivir con endometriosis: Una guía para recuperar el bienestarCalificación: 5 de 5 estrellas5/5 (5)
- Sana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónDe EverandSana tu Cuerpo, Calma tu Mente: Desintoxicar Hígado, Intestino Permeable, Salud Hormonal, Curación Emocional, Relajación, Ansiedad y Sanidad mental, Atención Plena, Psicoterapia y NutriciónCalificación: 5 de 5 estrellas5/5 (4)
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)De EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Calificación: 5 de 5 estrellas5/5 (9)
- Las Enfermedades comienzan y terminan en tu mente: Una guía para la autosanaciónDe EverandLas Enfermedades comienzan y terminan en tu mente: Una guía para la autosanaciónCalificación: 4 de 5 estrellas4/5 (4)
- Cuerpo Tóxico: Como Liberar Tu Cuerpo De Las Toxinas Externas E Internas, Y Evitar Asi Los Efectos De Los Radicales LibresDe EverandCuerpo Tóxico: Como Liberar Tu Cuerpo De Las Toxinas Externas E Internas, Y Evitar Asi Los Efectos De Los Radicales LibresCalificación: 5 de 5 estrellas5/5 (2)
- UF0085 - Diagnóstico estético facial y corporalDe EverandUF0085 - Diagnóstico estético facial y corporalCalificación: 4 de 5 estrellas4/5 (5)
- Cambiar el pasado: Superar las experiencias traumáticas con la terapia estratégicaDe EverandCambiar el pasado: Superar las experiencias traumáticas con la terapia estratégicaCalificación: 5 de 5 estrellas5/5 (4)
- Guía práctica para la refracción ocularDe EverandGuía práctica para la refracción ocularCalificación: 5 de 5 estrellas5/5 (2)
- Flores de Bach Recursos y estrategias terapéuticasDe EverandFlores de Bach Recursos y estrategias terapéuticasCalificación: 3.5 de 5 estrellas3.5/5 (3)
- Lactancia humana y fonoaudiología: Guía para madres lactantesDe EverandLactancia humana y fonoaudiología: Guía para madres lactantesCalificación: 5 de 5 estrellas5/5 (4)
- El Libro de la Dieta Antiinflamatoria: Plan de 14 días para Sanar el Sistema inmunológico y Sentirte Mejor que NuncaDe EverandEl Libro de la Dieta Antiinflamatoria: Plan de 14 días para Sanar el Sistema inmunológico y Sentirte Mejor que NuncaCalificación: 4.5 de 5 estrellas4.5/5 (14)
- Dieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasDe EverandDieta Para El Reflujo Biliar y Gastritis Alcalina - Incluye 20 Deliciosas Recetas Libres de Gluten y de Lácteos Para Tratar y Aliviar el Reflujo Biliar y Sus Molestos SíntomasCalificación: 4 de 5 estrellas4/5 (9)
- El código de la diabetes: Prevenir y revertir la diabetes tipo 2 de manera naturalDe EverandEl código de la diabetes: Prevenir y revertir la diabetes tipo 2 de manera naturalCalificación: 4.5 de 5 estrellas4.5/5 (24)