También podría gustarte
- Semiologia Sistema HematopoyeticoDocumento20 páginasSemiologia Sistema HematopoyeticoEsmeralda Quezada100% (8)
- Matematica para Los NegociosDocumento19 páginasMatematica para Los Negociosaa bbAún no hay calificaciones
- Implicacion LegalagraviosDocumento38 páginasImplicacion LegalagraviosGrace Vanesa LunaAún no hay calificaciones
- Morfologia Bucal CaninoDocumento9 páginasMorfologia Bucal CaninoGian Carlo Arroba RodriguezAún no hay calificaciones
- Modelo de Leavell y ClarkDocumento3 páginasModelo de Leavell y ClarkJulio NaizirAún no hay calificaciones
- SANGREDocumento5 páginasSANGREdaniel velazcoAún no hay calificaciones
- Etapas de La FiebreDocumento9 páginasEtapas de La FiebreyrkaAún no hay calificaciones
- Taller 2 de Molecular Enzimas de Restricción.Documento14 páginasTaller 2 de Molecular Enzimas de Restricción.MICHEL CAMILA GARZ?N PULIDOAún no hay calificaciones
- Inserto COVID 19 Ichroma II Con Guía PDFDocumento6 páginasInserto COVID 19 Ichroma II Con Guía PDFRoberto AriasAún no hay calificaciones
- Sustentabilidad en ChileDocumento10 páginasSustentabilidad en ChileJoel EstebanAún no hay calificaciones
- Saturación de OxigenoDocumento14 páginasSaturación de OxigenoAranza ArteagaAún no hay calificaciones
- Funciones Trigonometricas InversasDocumento6 páginasFunciones Trigonometricas InversasPedro A. Marrone G.Aún no hay calificaciones
- Lista de Precios de Medicamentos en BoliviaDocumento20 páginasLista de Precios de Medicamentos en BoliviaPágina SieteAún no hay calificaciones
- 28Documento47 páginas28Maximiliano Fabrizzio Fredes AbarzaAún no hay calificaciones
- Práctica 7. 2018-2Documento2 páginasPráctica 7. 2018-2Chessmaster98Aún no hay calificaciones
- Informe N°4 Morfología de HojaDocumento23 páginasInforme N°4 Morfología de Hojasamuel david borja martinezAún no hay calificaciones
- Manual Practicas Quimica IIDocumento121 páginasManual Practicas Quimica IIluisgutierres100% (1)
- Isomería Óptica y MedicamentosDocumento3 páginasIsomería Óptica y MedicamentosFerx Torres60% (5)
- Agentes Causantes de EnfermedadDocumento1 páginaAgentes Causantes de EnfermedadHugo Santiago Santiago100% (1)
- Informe Sobre Tipificación SanguineaDocumento8 páginasInforme Sobre Tipificación SanguineaTR María100% (1)
- Clasificación de Los Artrópodos de Importancia MédicaDocumento6 páginasClasificación de Los Artrópodos de Importancia Médicaangelika1211Aún no hay calificaciones
- Identificación de Azúcares y Almidones - LugolDocumento4 páginasIdentificación de Azúcares y Almidones - LugolLupita AlanAún no hay calificaciones
- EPR-Guías 1 Al 13 2017-I Química-I Ing. Industrial OkDocumento17 páginasEPR-Guías 1 Al 13 2017-I Química-I Ing. Industrial OkReynaldo CarreñoAún no hay calificaciones
- Resultados de Covid-19: NegativoDocumento1 páginaResultados de Covid-19: NegativoRene QUISPE LIRAAún no hay calificaciones
- Practica EstadistcaDocumento9 páginasPractica EstadistcaKenny SalazarAún no hay calificaciones
- Cultivos MicológicosDocumento7 páginasCultivos MicológicosKatherine Alexandra Merchan GalarzaAún no hay calificaciones
- DENGUEDocumento32 páginasDENGUEKaren PastorAún no hay calificaciones
- GuiaDocumento34 páginasGuiaSelena LovesGomezAún no hay calificaciones
- Caldo Clark y LubsDocumento1 páginaCaldo Clark y LubsOscar RamirezAún no hay calificaciones
- ACABADODocumento55 páginasACABADOJavier Rooque0% (1)
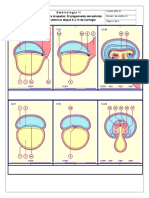
- Actividad Plegamiento Embrion para Maquetas de AcetatoDocumento4 páginasActividad Plegamiento Embrion para Maquetas de AcetatoXavier Rivera100% (1)
- Mapa Conceptual VacunasDocumento1 páginaMapa Conceptual VacunasNatalia SuárezAún no hay calificaciones
- UnidoestadisticaDocumento63 páginasUnidoestadisticaYuliana Gabriela González LópezAún no hay calificaciones
- Adn MonografiaDocumento48 páginasAdn MonografiaLuis Arnaldo Meza Guerrero100% (1)
- Antianémicos y Otros HematínicosDocumento13 páginasAntianémicos y Otros HematínicosEsperanza Silva YeroAún no hay calificaciones
- Banqueo Ec2-Quimica OrganicaDocumento25 páginasBanqueo Ec2-Quimica Organicaporcelains dollAún no hay calificaciones
- Inducción de Seguridad - Softys - Contratistas 2021 Rev02.Documento75 páginasInducción de Seguridad - Softys - Contratistas 2021 Rev02.valeskajaAún no hay calificaciones
- Examen Final G1 MicrobiologiaDocumento3 páginasExamen Final G1 Microbiologiakathy.cc96100% (1)
- Fisicoqmca Lab.2Documento6 páginasFisicoqmca Lab.2ALFREDO MAMANI CALLEAún no hay calificaciones
- Partes Del MicroscopioDocumento2 páginasPartes Del MicroscopioRobert Cristofer Ortiz LuquillasAún no hay calificaciones
- Caso Clínico TalasemiaDocumento4 páginasCaso Clínico TalasemiaMei MeiAún no hay calificaciones
- Proceso ADME Jazmín ChasipantaDocumento2 páginasProceso ADME Jazmín ChasipantaJazmin Chasipanta50% (2)
- Qca Semana 7Documento4 páginasQca Semana 7Jorge Antonio Loza CachayAún no hay calificaciones
- Taller Teorico - Practico de VirologiaDocumento5 páginasTaller Teorico - Practico de VirologiaANGELA NICOLE REINA BARBOSA100% (1)
- PRÁCTICA 1 Exámen Preliminar y Análisis ElementalDocumento8 páginasPRÁCTICA 1 Exámen Preliminar y Análisis ElementalAC Danika100% (1)
- Adn SateliteDocumento5 páginasAdn SateliteJhon Pendola LoarteAún no hay calificaciones
- Sal Yodada Datos de SeguridadDocumento1 páginaSal Yodada Datos de SeguridadKris Paola100% (1)
- Mapa de Metodos AnticonceptivosDocumento1 páginaMapa de Metodos AnticonceptivosIvonne PérezAún no hay calificaciones
- Ejercicios de Nomenclatura de Cadenas RamificadasDocumento1 páginaEjercicios de Nomenclatura de Cadenas RamificadasJuan JoseAún no hay calificaciones
- Soluciones Química - QuímicaDocumento5 páginasSoluciones Química - QuímicaDorisNoeliaNúñezBeniqueAún no hay calificaciones
- Ejercicios-de-Probabilidad PaoDocumento18 páginasEjercicios-de-Probabilidad Paochapita1086Aún no hay calificaciones
- Clasificacion de Las Formas Farmaceuticas GaseosasDocumento1 páginaClasificacion de Las Formas Farmaceuticas GaseosasDany OscoAún no hay calificaciones
- Historia Natural de Fiebre de La LASSADocumento15 páginasHistoria Natural de Fiebre de La LASSAElsa Chalé CanAún no hay calificaciones
- DengueDocumento2 páginasDengueDaniel AriasAún no hay calificaciones
- Señalizaciones en El LaboratorioDocumento8 páginasSeñalizaciones en El LaboratorioraulAún no hay calificaciones
- Informe 10Documento8 páginasInforme 10LINA MARIA MOSQUERA NINCO100% (1)
- Exposición de Serología Líquido CefalorraquídeoDocumento23 páginasExposición de Serología Líquido CefalorraquídeoRoberto Alonso0% (1)
- Informe de Baño de MariaDocumento4 páginasInforme de Baño de MariaStivens HenaoAún no hay calificaciones
- Biologia ChargaffDocumento2 páginasBiologia ChargaffJenny CarboAún no hay calificaciones
- AGA Disturbios Respiratorios PLUS MEDIC ADocumento8 páginasAGA Disturbios Respiratorios PLUS MEDIC AGretchen LavadoAún no hay calificaciones
- Apuntes Todos Real Final 2022-09-08 20 - 50 - 42Documento64 páginasApuntes Todos Real Final 2022-09-08 20 - 50 - 42AlondraAún no hay calificaciones
- BicompartimentalDocumento27 páginasBicompartimentalfarmacia bioquimica 2016Aún no hay calificaciones
- Bicompartimental FarmacologiaDocumento20 páginasBicompartimental Farmacologiakyasarin12Aún no hay calificaciones
- Talleres Farmacologia 2023Documento39 páginasTalleres Farmacologia 2023Esmeralda QuezadaAún no hay calificaciones
- Resumen Post MortemDocumento6 páginasResumen Post MortemEsmeralda QuezadaAún no hay calificaciones
- Cuestionario: Laboratorio Toxicología Clínica y Forense Farm 802Documento11 páginasCuestionario: Laboratorio Toxicología Clínica y Forense Farm 802Esmeralda QuezadaAún no hay calificaciones
- ENDOCRINO ArgenteDocumento14 páginasENDOCRINO ArgenteEsmeralda QuezadaAún no hay calificaciones
- Resumen Semiología GeneralDocumento25 páginasResumen Semiología GeneralEsmeralda QuezadaAún no hay calificaciones
- Gine ArgenteDocumento4 páginasGine ArgenteEsmeralda QuezadaAún no hay calificaciones
- Clase 9 - Modelo BicompartimentalDocumento19 páginasClase 9 - Modelo BicompartimentalEsmeralda QuezadaAún no hay calificaciones
- BIOL260 Clase 03 Actividad Enzimática y Mecanismos de RegulaciónDocumento60 páginasBIOL260 Clase 03 Actividad Enzimática y Mecanismos de RegulaciónEsmeralda QuezadaAún no hay calificaciones
- Complejo Multienzimatico de La ReplicacionDocumento2 páginasComplejo Multienzimatico de La ReplicacionEsmeralda QuezadaAún no hay calificaciones
- Resumen Osteocarilaginoso EDocumento4 páginasResumen Osteocarilaginoso EEsmeralda QuezadaAún no hay calificaciones
- Guia No 4 Criterios de Reactividad PDFDocumento1 páginaGuia No 4 Criterios de Reactividad PDFEsmeralda QuezadaAún no hay calificaciones
- Guia No 3 Resonancia PDFDocumento1 páginaGuia No 3 Resonancia PDFEsmeralda QuezadaAún no hay calificaciones
- Sintesis de Cadena RetrasadaDocumento2 páginasSintesis de Cadena RetrasadaEsmeralda QuezadaAún no hay calificaciones
- Ejercicio Bolus 2019Documento2 páginasEjercicio Bolus 2019Esmeralda Quezada100% (1)
- Entregable 3 M1Documento4 páginasEntregable 3 M1Esmeralda QuezadaAún no hay calificaciones
- ALGUNAS PREGUNTAS TIPOS III SolemneDocumento3 páginasALGUNAS PREGUNTAS TIPOS III SolemneEsmeralda QuezadaAún no hay calificaciones
- Spab111 FinalDocumento3 páginasSpab111 FinalEsmeralda Quezada0% (1)
- Actividad Integradora 3algebrando La VidaDocumento4 páginasActividad Integradora 3algebrando La VidaVictor HernándezAún no hay calificaciones
- Informe Balance de Materia de Mermelada de UvaDocumento5 páginasInforme Balance de Materia de Mermelada de UvaJoel Paucar Sinche100% (1)
- Pensamiento Algoritmico Escenario 8Documento7 páginasPensamiento Algoritmico Escenario 8Äłějąņđřø MåľăğøņAún no hay calificaciones
- PDFDocumento181 páginasPDFjhony1523Aún no hay calificaciones
- Cuestionario 2 Algebra Lineal Matrices PDFDocumento7 páginasCuestionario 2 Algebra Lineal Matrices PDFAngel CaminoAún no hay calificaciones
- Manual TX30ESDocumento14 páginasManual TX30ESCristobal JimenezAún no hay calificaciones
- 4 ElectroneumáticoDocumento8 páginas4 Electroneumáticojuan esteban marinAún no hay calificaciones
- Hoja de Procesos Practica de TornoDocumento4 páginasHoja de Procesos Practica de TornoJesus SerranoAún no hay calificaciones
- Practica - 06 - Razonamiento - Matematico - Sin Claves - EstudiantesDocumento7 páginasPractica - 06 - Razonamiento - Matematico - Sin Claves - EstudiantesKati MamaniAún no hay calificaciones
- Dimensiones Fancoils Del 1 Hasta 5ton SEER 20.5Documento51 páginasDimensiones Fancoils Del 1 Hasta 5ton SEER 20.5Jason RojasAún no hay calificaciones
- Correos Electrónicos 101859055-0e1cap-7-Integrales-Sobre-Trayectorias-y-Superficies PDFDocumento77 páginasCorreos Electrónicos 101859055-0e1cap-7-Integrales-Sobre-Trayectorias-y-Superficies PDFEdgar Ernesto Granados Betancourt100% (1)
- Amplificador de Instrumentacion - SensoricxDocumento7 páginasAmplificador de Instrumentacion - SensoricxBrain Magdaleno DazaAún no hay calificaciones
- Problemas02 PDFDocumento3 páginasProblemas02 PDFRuddy Alfredo Cabrejos RamosAún no hay calificaciones
- Nomenclatura Química InorgánicaDocumento55 páginasNomenclatura Química InorgánicaAlejandra Isabel Hinojosa RiosAún no hay calificaciones
- Practica de Laboratorio Caida Libre Movimiento Rectilineo Uniforme y AceleeradoDocumento14 páginasPractica de Laboratorio Caida Libre Movimiento Rectilineo Uniforme y AceleeradoFabian HigueraAún no hay calificaciones
- Unidad 1Documento63 páginasUnidad 1RobertLopezFudoAún no hay calificaciones
- Control and Monitoring Online of A Dryer For EsDocumento5 páginasControl and Monitoring Online of A Dryer For EsAlejandro Esteban Ramírez NovoaAún no hay calificaciones
- MRUVDocumento16 páginasMRUVFettuccini EstropajoAún no hay calificaciones
- Manual de Operacion y Mantenimiento AguaDocumento14 páginasManual de Operacion y Mantenimiento AguaMailerNereoSalvadorLugoAún no hay calificaciones
- Análisis de VigasDocumento7 páginasAnálisis de VigasFrancy LeonAún no hay calificaciones
- Métodos de RemediacionDocumento25 páginasMétodos de RemediacionAnonymous 666Aún no hay calificaciones
- HistologiaDocumento32 páginasHistologiaSalvador Antonio Caldera CamachoAún no hay calificaciones
- (r5) Lenguaje-Reconocimiento de Categorías Gramaticales-Prof - Christian Linares-AsmDocumento39 páginas(r5) Lenguaje-Reconocimiento de Categorías Gramaticales-Prof - Christian Linares-AsmALLISON KARINA SANTOS BOBADILLAAún no hay calificaciones
- Lab Nro 2 ELT 279Documento4 páginasLab Nro 2 ELT 279Alejandro Quinteros CabreraAún no hay calificaciones
- Algunas Fisiopatias de Frutos, Tallos y Hojas en Cultivos Protegidos PDFDocumento15 páginasAlgunas Fisiopatias de Frutos, Tallos y Hojas en Cultivos Protegidos PDFGeorgette MuñozAún no hay calificaciones
- Armen InformeDocumento9 páginasArmen InformeYuriko GoicocheaAún no hay calificaciones
- Memoria de Calculo de Poblaciones FuturasDocumento6 páginasMemoria de Calculo de Poblaciones FuturasyelsitoAún no hay calificaciones
- V. Regresión Logística y Regresión de PoissonDocumento29 páginasV. Regresión Logística y Regresión de PoissonJoseLuisLópezAún no hay calificaciones