También podría gustarte
- Fisicoquímica II - Coeficiente de Actividad de Un ElectrolitoDocumento14 páginasFisicoquímica II - Coeficiente de Actividad de Un ElectrolitoDiego Vergaray D'Arrigo100% (1)
- Medicion y Mejoramiento de La ProductividadDocumento55 páginasMedicion y Mejoramiento de La ProductividadMathias Leiva100% (7)
- Complejo Mayor de HistocompatibilidadDocumento4 páginasComplejo Mayor de HistocompatibilidadANGELINE STEFANIA CARBONEL CORNEJOAún no hay calificaciones
- Numeros en Qeqchi de 1 Al 100Documento1 páginaNumeros en Qeqchi de 1 Al 100sonyAún no hay calificaciones
- Triangulación en Analisis de ContenidoDocumento16 páginasTriangulación en Analisis de ContenidoMarinely Oviedo100% (1)
- Propiedades de Sustancias PurasDocumento24 páginasPropiedades de Sustancias PurasCrispeta YerryAún no hay calificaciones
- Tesis Doctoral Humor 2015Documento305 páginasTesis Doctoral Humor 2015Marinely OviedoAún no hay calificaciones
- Reporte TermoquimicaDocumento8 páginasReporte TermoquimicaDiianaLauraMelendezAún no hay calificaciones
- Reporte Diagrama Binario Solido LiquidoDocumento28 páginasReporte Diagrama Binario Solido LiquidoErika AyalaAún no hay calificaciones
- Reacción de Identificación de Cationes Grupo IIDocumento1 páginaReacción de Identificación de Cationes Grupo IIJoel PantojaAún no hay calificaciones
- CAPITULO I VOLUMETRÍA - Q.A Cuantitativa Bromatologia - S2 - 2022Documento11 páginasCAPITULO I VOLUMETRÍA - Q.A Cuantitativa Bromatologia - S2 - 2022Marco Antonio Sicard arceAún no hay calificaciones
- Práctica 2. DETERMINACIÓN DE SULFATOS PDFDocumento14 páginasPráctica 2. DETERMINACIÓN DE SULFATOS PDFfrancia.castilloAún no hay calificaciones
- Informe 2 Reaciones Quimica 2020Documento12 páginasInforme 2 Reaciones Quimica 2020Sergio SusaraAún no hay calificaciones
- EVAPORACIÓNDocumento20 páginasEVAPORACIÓNYeferson Andy Alexis Chuchon GomezAún no hay calificaciones
- QMC 104 Lab 7Documento21 páginasQMC 104 Lab 7Cruz Torrez Jhenny MichelAún no hay calificaciones
- E2208004 6 Taller Smog FotoquimicoDocumento8 páginasE2208004 6 Taller Smog FotoquimicoRaul GranadosAún no hay calificaciones
- Informe 3 Trayectoria ParabólicaDocumento5 páginasInforme 3 Trayectoria ParabólicaRicardo JesusAún no hay calificaciones
- Calculo Diferencial - Listo para EnviarDocumento23 páginasCalculo Diferencial - Listo para EnviardiegoAún no hay calificaciones
- Laboratorio de Fisica II-UNMSMDocumento16 páginasLaboratorio de Fisica II-UNMSMPablo PachecoAún no hay calificaciones
- PRACTICA 2. Estandarización de HCL y NaOH.Documento4 páginasPRACTICA 2. Estandarización de HCL y NaOH.Sofia Paredes OrtizAún no hay calificaciones
- Comp1 Dismutación Ag - NH3 EZPDocumento2 páginasComp1 Dismutación Ag - NH3 EZPRafael GonzálezAún no hay calificaciones
- Laboratorio Analítica Práctica 3Documento11 páginasLaboratorio Analítica Práctica 3Ethel DekkerAún no hay calificaciones
- Practica #03 Modelos Moleculares e HibridaciónDocumento5 páginasPractica #03 Modelos Moleculares e HibridaciónAlexander VarasAún no hay calificaciones
- Reporte Post-Laboratorio. Práctica 6Documento5 páginasReporte Post-Laboratorio. Práctica 6KarenAún no hay calificaciones
- Reporte de Curvas de Calibracion Cobre y NiquelDocumento9 páginasReporte de Curvas de Calibracion Cobre y NiquelOliver JinZo MonzalvoAún no hay calificaciones
- Reacciones de Transposición (Rearreglos) JPLLDocumento21 páginasReacciones de Transposición (Rearreglos) JPLLAdrian Camilo Mariño AldanaAún no hay calificaciones
- Benemérita Universidad Autónoma de Puebla Practica 3Documento10 páginasBenemérita Universidad Autónoma de Puebla Practica 3Diego Armando Frias VazquezAún no hay calificaciones
- Dependencia de La Fem Con TDocumento17 páginasDependencia de La Fem Con Tgisela2592Aún no hay calificaciones
- Informe 2 Qca FisicaDocumento5 páginasInforme 2 Qca FisicaMatilde Santibáñez MéndezAún no hay calificaciones
- Primer Serie Vectores BQDDocumento4 páginasPrimer Serie Vectores BQDSebastian Torres SanchezAún no hay calificaciones
- 2022 Módulo Sobre Soluciones Electrolíticas y Soluciones No ElectrolíticasDocumento59 páginas2022 Módulo Sobre Soluciones Electrolíticas y Soluciones No ElectrolíticasGislena CaballeroAún no hay calificaciones
- Aminas y DerivadosDocumento55 páginasAminas y DerivadosDiego MorinigoAún no hay calificaciones
- Benzofenona DifenilaminaDocumento5 páginasBenzofenona DifenilaminaArenita GuerronAún no hay calificaciones
- Informe Equilibrio Quimico LCHATELIERDocumento24 páginasInforme Equilibrio Quimico LCHATELIERjhofreAún no hay calificaciones
- Informe Conservacion de La EnergiaDocumento6 páginasInforme Conservacion de La Energiaanon_826658038Aún no hay calificaciones
- Guia de Laboratorio 5 Quimica OrganicaDocumento11 páginasGuia de Laboratorio 5 Quimica OrganicaJanice RomeroAún no hay calificaciones
- Informe 3 ValeriaDocumento22 páginasInforme 3 ValeriaTobi BerrospiAún no hay calificaciones
- Diagrama P&id DialisisDocumento3 páginasDiagrama P&id DialisisPaola Mamani TayaAún no hay calificaciones
- Tabla de DisolventesDocumento13 páginasTabla de DisolventesAldo100% (2)
- Calculos Lab 11Documento17 páginasCalculos Lab 11JherssyAún no hay calificaciones
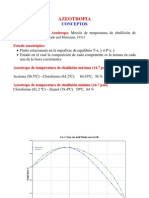
- AzeotropiaDocumento25 páginasAzeotropiaRicardo KobainAún no hay calificaciones
- Cblancot - Lab 3. Descenso en El Punto de CongelaciónDocumento10 páginasCblancot - Lab 3. Descenso en El Punto de CongelaciónNataliaRiveroArenas100% (1)
- Informe N-5Documento4 páginasInforme N-5miguel concha ccanchiAún no hay calificaciones
- Práctica 3 Laboratorio de Equilibrio y CineticaDocumento4 páginasPráctica 3 Laboratorio de Equilibrio y CineticaDany PandaAún no hay calificaciones
- T.P N°8 Determinación de Cloruros Por Método Volhard (Titulación Por Retorno)Documento2 páginasT.P N°8 Determinación de Cloruros Por Método Volhard (Titulación Por Retorno)Triz MaruAún no hay calificaciones
- Química Ambiental IDocumento13 páginasQuímica Ambiental IVilchis Barajas UrielAún no hay calificaciones
- Cuestionario y Diagrama de FlujoDocumento7 páginasCuestionario y Diagrama de FlujoLeonardo OrtizAún no hay calificaciones
- Producto Solubilidad Hidroxido de BarioDocumento8 páginasProducto Solubilidad Hidroxido de BarioIvan IvenianAún no hay calificaciones
- Hibridacion y Modelo - Duran - Villeda - OsminDocumento3 páginasHibridacion y Modelo - Duran - Villeda - OsminOSMIN DURAN VILLEDAAún no hay calificaciones
- DEBORA JIMENA MENDOZA SILVA - Tarea 3 Del Curso de FISICOQUÍMICA A IIDocumento5 páginasDEBORA JIMENA MENDOZA SILVA - Tarea 3 Del Curso de FISICOQUÍMICA A IIÁlvaro Olórtegui IglesiasAún no hay calificaciones
- Sistemas de Tres LíquidosDocumento8 páginasSistemas de Tres LíquidosevelinAún no hay calificaciones
- Determinación Curvas de La Solubilidad (2020)Documento2 páginasDeterminación Curvas de La Solubilidad (2020)Helber Dario Melo FigueroaAún no hay calificaciones
- Informe 8 Físico QuímicaDocumento6 páginasInforme 8 Físico QuímicaBrayan Alexis0% (1)
- 1er Informe de FisicoquimicaDocumento34 páginas1er Informe de FisicoquimicaDiego Gavino RomeroAún no hay calificaciones
- Organizador Gráfico de Análisis InstrumentalDocumento1 páginaOrganizador Gráfico de Análisis InstrumentalANA CECILIA MALDONADO MORINAún no hay calificaciones
- RecristalizaciónDocumento10 páginasRecristalizaciónKatherin ChapidAún no hay calificaciones
- Apuntes - Hidrólisis y S. AmortiguadorasDocumento10 páginasApuntes - Hidrólisis y S. AmortiguadoraskierkegardAún no hay calificaciones
- ManLabQA2 (Q) 2020-1 Parte2 PDFDocumento43 páginasManLabQA2 (Q) 2020-1 Parte2 PDFPedro HernandezAún no hay calificaciones
- Laboratorio de Mecanica 4Documento12 páginasLaboratorio de Mecanica 4David RodriguezAún no hay calificaciones
- Tarea 8 - SolucionarioDocumento39 páginasTarea 8 - SolucionarioFabritzio- KunAún no hay calificaciones
- Determinación de Hierro Por UV-visDocumento6 páginasDeterminación de Hierro Por UV-visyeisirhetguerrero07Aún no hay calificaciones
- Titulación Potenciométrica de Especies ÁcidoDocumento10 páginasTitulación Potenciométrica de Especies ÁcidoJhonAún no hay calificaciones
- Marco Teorico de La Segunda LeyDocumento11 páginasMarco Teorico de La Segunda LeyMarck Andree Limaymanta PardaveAún no hay calificaciones
- RT TermoDocumento150 páginasRT TermoiretchaiAún no hay calificaciones
- Temas Selectos de Quimica 3er Parcial BachilleratoDocumento35 páginasTemas Selectos de Quimica 3er Parcial BachilleratoSmuert Poot VazquezAún no hay calificaciones
- Historia de La Propaganda - Alejandro Pizarroso Quintero PDFDocumento239 páginasHistoria de La Propaganda - Alejandro Pizarroso Quintero PDFMarinely Oviedo83% (6)
- Tesis Eduardo Pateiro PDFDocumento311 páginasTesis Eduardo Pateiro PDFMarinely OviedoAún no hay calificaciones
- QUIPU Revista Científica PNFCP UPTAEB Año 1 #1 Noviembre 2016Documento126 páginasQUIPU Revista Científica PNFCP UPTAEB Año 1 #1 Noviembre 2016Marinely OviedoAún no hay calificaciones
- RRPP y Marketing PDFDocumento13 páginasRRPP y Marketing PDFMarinely OviedoAún no hay calificaciones
- Transcomplejidad GerenciaDocumento7 páginasTranscomplejidad GerenciaMarinely OviedoAún no hay calificaciones
- AcentoDocumento38 páginasAcentoEdgar RodriguezAún no hay calificaciones
- Metodo CelosiaDocumento4 páginasMetodo CelosiaDiego Riedemann100% (1)
- Tema3.PDF Campo ElectricoDocumento35 páginasTema3.PDF Campo ElectricoIanel Mayta LovatonAún no hay calificaciones
- El Registro ContableDocumento5 páginasEl Registro ContableAlvaroAún no hay calificaciones
- 2 Examen Oft Imp 2018 1Documento4 páginas2 Examen Oft Imp 2018 1Fanny Ozuna VillalvaAún no hay calificaciones
- Completos Apuntes MercantilDocumento113 páginasCompletos Apuntes MercantilElena CabanzónAún no hay calificaciones
- Disciplina JaponesaDocumento6 páginasDisciplina JaponesaFatima ArmasAún no hay calificaciones
- Caso de ÉxitoDocumento13 páginasCaso de ÉxitocenebaAún no hay calificaciones
- Presentación Educativa Diapositivas para Proyecto de Educación Coloridas Rosa, Blanco y VerdeDocumento17 páginasPresentación Educativa Diapositivas para Proyecto de Educación Coloridas Rosa, Blanco y VerdeJesus García ReyesAún no hay calificaciones
- Muestreo PoblacionalDocumento22 páginasMuestreo PoblacionalNestor Alejandro Velazco SilvaAún no hay calificaciones
- Manual EdiproDocumento31 páginasManual EdiprocooterAún no hay calificaciones
- GLAUCOMADocumento61 páginasGLAUCOMACarlitos AyalaAún no hay calificaciones
- Metrado Edificio Trujillo-Constructora Baldera E.I.R.LDocumento5 páginasMetrado Edificio Trujillo-Constructora Baldera E.I.R.LAndherson Junior GAún no hay calificaciones
- 636 0896398sa319Documento27 páginas636 0896398sa319Julieth PrietoAún no hay calificaciones
- Carta Al Estudiante TeoríaDocumento3 páginasCarta Al Estudiante TeoríaIsberto MartínezAún no hay calificaciones
- Guía 1 Mate FinancieraDocumento9 páginasGuía 1 Mate FinancieraMatias GinerAún no hay calificaciones
- Zonificacion y Codificacion CuencasDocumento76 páginasZonificacion y Codificacion Cuencasivan890720Aún no hay calificaciones
- Ficha de Registro de Sitios Con Arte Rupestre y Piedras Con CúpulasDocumento6 páginasFicha de Registro de Sitios Con Arte Rupestre y Piedras Con CúpulasMario A. Ramos0% (1)
- AnestesiaDocumento84 páginasAnestesiaALFREDO RODRIGUEZ DEL ANGELAún no hay calificaciones
- Dureza Del Agua Potable en ChileDocumento5 páginasDureza Del Agua Potable en ChileEduardo Luis Venegas ObandoAún no hay calificaciones
- Gestion de UnicelDocumento51 páginasGestion de UnicelAriadna Montiel BolanAún no hay calificaciones
- 9 de EneroDocumento2 páginas9 de EneroEdgar MontecerAún no hay calificaciones
- MFPH II Plan de Clase 12Documento9 páginasMFPH II Plan de Clase 12Pedro Black Diamontt100% (1)
- Escalas de TemperaturaDocumento1 páginaEscalas de Temperaturasayani lopezAún no hay calificaciones
- 2182-Texto Del Artículo-6791-1-10-20161027Documento26 páginas2182-Texto Del Artículo-6791-1-10-20161027toleAún no hay calificaciones
- Plan de Trabajo para La Actividad Denominada: ": Memoria DescriptivaDocumento9 páginasPlan de Trabajo para La Actividad Denominada: ": Memoria DescriptivaEmerson Crisostomo LaurenteAún no hay calificaciones
- Unmsm sabadoqqFLsrjXbGKFDocumento5 páginasUnmsm sabadoqqFLsrjXbGKFElizabeth Cristina Asencios ToledoAún no hay calificaciones
- Temario Lesiones (Manuel)Documento17 páginasTemario Lesiones (Manuel)Javier Estelles MuñozAún no hay calificaciones
- Informe Gerencial PDFDocumento14 páginasInforme Gerencial PDFMilena Castro BermudezAún no hay calificaciones