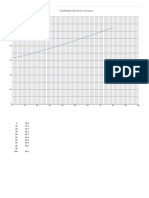
También podría gustarte
- Análisis de circuitos eléctricos Estado estableDe EverandAnálisis de circuitos eléctricos Estado estableCalificación: 5 de 5 estrellas5/5 (8)
- Análisis de circuitos eléctricos. Un enfoque teóricoDe EverandAnálisis de circuitos eléctricos. Un enfoque teóricoAún no hay calificaciones
- Análisis y diseño de circuitos eléctricos: Teoría y prácticaDe EverandAnálisis y diseño de circuitos eléctricos: Teoría y prácticaAún no hay calificaciones
- (Schaum) W G MacLean - E W Nelson - Teoria y Problemas de Mecánica Técnica - Estática y Dinámica (1971, McGraw-Hill) PDFDocumento361 páginas(Schaum) W G MacLean - E W Nelson - Teoria y Problemas de Mecánica Técnica - Estática y Dinámica (1971, McGraw-Hill) PDFRichard Balseca100% (3)
- Introducción al análisis de supervivencia avanzadaDe EverandIntroducción al análisis de supervivencia avanzadaAún no hay calificaciones
- Introducción a la matemática discretaDe EverandIntroducción a la matemática discretaCalificación: 5 de 5 estrellas5/5 (1)
- Química general para las ciencias ambientalesDe EverandQuímica general para las ciencias ambientalesAún no hay calificaciones
- Manual Alarma Home SafeDocumento8 páginasManual Alarma Home SafeCarlos AnibalAún no hay calificaciones
- Manual Ecg Fukudac c120Documento43 páginasManual Ecg Fukudac c120Miguel Figueroa67% (3)
- BateriasDocumento35 páginasBateriasAlex GarciaAún no hay calificaciones
- Leyes Ambientales de El SalvadorDocumento15 páginasLeyes Ambientales de El SalvadorSandra Rodriguez38% (8)
- El problema de complementariedad No lineal: Teoría, aplicaciones y nuevos algoritmos para su soluciónDe EverandEl problema de complementariedad No lineal: Teoría, aplicaciones y nuevos algoritmos para su soluciónAún no hay calificaciones
- Ecuaciones Diferenciales para IngenierosDocumento256 páginasEcuaciones Diferenciales para IngenierosCristian Rojas100% (2)
- La integral de Lebesgue en RN: Teoría y problemasDe EverandLa integral de Lebesgue en RN: Teoría y problemasAún no hay calificaciones
- PDFDocumento109 páginasPDFHenry MolinaAún no hay calificaciones
- Física EstadisticaDocumento103 páginasFísica EstadisticacyanhidricoAún no hay calificaciones
- PDFDocumento2 páginasPDFnayabimtiaz1997Aún no hay calificaciones
- La Termodinamica de Membranas Artificiales Bajo La Influencia de Moleculas Polares y No PolaresDocumento85 páginasLa Termodinamica de Membranas Artificiales Bajo La Influencia de Moleculas Polares y No PolaresSofia Isabella GuevaraAún no hay calificaciones
- Dinamica Celulas Cancerigenas Vs Sistema InmunologicoDocumento110 páginasDinamica Celulas Cancerigenas Vs Sistema InmunologicoLuis Fernando Granados BorjaAún no hay calificaciones
- MemoriaDocumento37 páginasMemoriaGeraldine CastilloAún no hay calificaciones
- Carlosandresplazasriano 2011Documento77 páginasCarlosandresplazasriano 2011yenny monsalveAún no hay calificaciones
- Elementos de La Cohomologı́a de de Rham Desde La Teorı́a de FibradosDocumento52 páginasElementos de La Cohomologı́a de de Rham Desde La Teorı́a de FibradosCarlos Francisco Flores GaliciaAún no hay calificaciones
- Inversacion Electronica-Mecatronica Parte1Documento20 páginasInversacion Electronica-Mecatronica Parte1Juddas KainAún no hay calificaciones
- Blanco Guillén, María Montaña TFGDocumento174 páginasBlanco Guillén, María Montaña TFGreggaebgtaAún no hay calificaciones
- CONICET Digital Nro. ADocumento123 páginasCONICET Digital Nro. Ajordyvele777Aún no hay calificaciones
- 03 Tres ColetivosDocumento56 páginas03 Tres ColetivosKevin GuilletAún no hay calificaciones
- Julio Cesar Godines Delgado 2019Documento155 páginasJulio Cesar Godines Delgado 2019Mario Hilari CalderonAún no hay calificaciones
- Conceptos Básicos Cúantica 1Documento54 páginasConceptos Básicos Cúantica 1Cristin LeeAún no hay calificaciones
- GRL ThesisDocumento250 páginasGRL ThesisOsayomwanbor Ehi-EgharevbaAún no hay calificaciones
- PedrazaMartin PublicoDocumento296 páginasPedrazaMartin Publicobarreaza_1Aún no hay calificaciones
- 371MC JNJDocumento111 páginas371MC JNJLEE ALVAREZAún no hay calificaciones
- Tesis-Reducción de La Dimensionalidad de Microarreglos ADN para Estimar La Función de Supervivencia - Edgar Alamilla Jiménez - UJATDocumento78 páginasTesis-Reducción de La Dimensionalidad de Microarreglos ADN para Estimar La Función de Supervivencia - Edgar Alamilla Jiménez - UJATEdgar Alamilla JiménezAún no hay calificaciones
- TFM - JUAREZ MARTINEZ - Modelos Dinámicos en Biología y EconomíaDocumento94 páginasTFM - JUAREZ MARTINEZ - Modelos Dinámicos en Biología y EconomíaManayay WilmerAún no hay calificaciones
- Introduccion A La Teoria de Codigos - Codigos Ciclicos.Documento53 páginasIntroduccion A La Teoria de Codigos - Codigos Ciclicos.Jhovanny CondoriAún no hay calificaciones
- El Rompecabezas Del Adn - 7-12Documento7 páginasEl Rompecabezas Del Adn - 7-12Mayra HernandezAún no hay calificaciones
- Pre Tesis EjemploDocumento45 páginasPre Tesis EjemploBryan Asto CamposAún no hay calificaciones
- Simulaciones de FlujoDocumento266 páginasSimulaciones de FlujoEliizabeth SFAún no hay calificaciones
- Variable Compleja - Artemio Gonzalez Lopez PDFDocumento62 páginasVariable Compleja - Artemio Gonzalez Lopez PDFCristhian FFFAún no hay calificaciones
- Patrones de Turing en Sistemas Biologicos - Ledesma-DA-Tesis PDFDocumento124 páginasPatrones de Turing en Sistemas Biologicos - Ledesma-DA-Tesis PDFVicenteAún no hay calificaciones
- Metodos Numericos Por Cadenas de MarkovDocumento104 páginasMetodos Numericos Por Cadenas de MarkovMiguel Angel Varela LópezAún no hay calificaciones
- Tesis X3043601Documento157 páginasTesis X3043601julioAún no hay calificaciones
- MATRICESDocumento149 páginasMATRICESFabrizzio Reyes M100% (2)
- Instituto Politécnico Nacional: Escuela Superior de Física y MatemáticasDocumento76 páginasInstituto Politécnico Nacional: Escuela Superior de Física y MatemáticasManuel Gael Camara NacerAún no hay calificaciones
- Dinamica Molecular y Met Monte CarloDocumento20 páginasDinamica Molecular y Met Monte CarloRichard PachacamaAún no hay calificaciones
- Distribución MacrocanónicaDocumento22 páginasDistribución MacrocanónicaJose ramon cornelio torresAún no hay calificaciones
- Gec 2 Estructuras MicrobianasDocumento8 páginasGec 2 Estructuras MicrobianasMENDOZA COLONIA CAMILO JOSEAún no hay calificaciones
- TFG EloyOrdizLeraDocumento95 páginasTFG EloyOrdizLerajaceAún no hay calificaciones
- Guia de Problemas Veterinaria 2023Documento23 páginasGuia de Problemas Veterinaria 2023Sofia SirrAún no hay calificaciones
- CopulasDocumento104 páginasCopulasJesus Enrique Miranda BlancoAún no hay calificaciones
- Bifurcaciones de Codimension Uno en Sistemas Discretos - Maria MataDocumento80 páginasBifurcaciones de Codimension Uno en Sistemas Discretos - Maria MataJOSE LUISAún no hay calificaciones
- Libro Sambarino HiperbolicidadDocumento134 páginasLibro Sambarino HiperbolicidadGerman ChiappeAún no hay calificaciones
- Teorema de Whitehead y Aproximacion CelularDocumento61 páginasTeorema de Whitehead y Aproximacion CelularNacho Lopez RomeroAún no hay calificaciones
- Tesis - Lic Complejos Simpliciales y Teoría de MorseDocumento97 páginasTesis - Lic Complejos Simpliciales y Teoría de MorseDiegoAún no hay calificaciones
- Mamani - Análisis Semántico Latente Mediante Descomposición en Valores SingularesDocumento44 páginasMamani - Análisis Semántico Latente Mediante Descomposición en Valores SingularesErnesto CortesAún no hay calificaciones
- Manual CTO - GenéticaDocumento19 páginasManual CTO - GenéticaKurai100% (7)
- La interacción del neonato: un instrumento de evaluación observacional, CITMI-NBDe EverandLa interacción del neonato: un instrumento de evaluación observacional, CITMI-NBAún no hay calificaciones
- Practica No.5Documento7 páginasPractica No.5Moy AguilarAún no hay calificaciones
- Practica No. 4Documento6 páginasPractica No. 4Moy AguilarAún no hay calificaciones
- Practica No.3Documento6 páginasPractica No.3Moy AguilarAún no hay calificaciones
- Aguilar Delgado Moises - ResumenDocumento1 páginaAguilar Delgado Moises - ResumenMoy AguilarAún no hay calificaciones
- Ctividad 7.1 - Aguilar Delgado MoisesDocumento9 páginasCtividad 7.1 - Aguilar Delgado MoisesMoy AguilarAún no hay calificaciones
- El Romanticismo en La ModernidadDocumento1 páginaEl Romanticismo en La ModernidadMoy AguilarAún no hay calificaciones
- Actividad 1.2aaguilar Delgado MoisesDocumento4 páginasActividad 1.2aaguilar Delgado MoisesMoy AguilarAún no hay calificaciones
- Actividad 2.1 Aguilar DelgadoDocumento3 páginasActividad 2.1 Aguilar DelgadoMoy AguilarAún no hay calificaciones
- Actividad 1.4aaguilar Delgado MoisesDocumento5 páginasActividad 1.4aaguilar Delgado MoisesMoy AguilarAún no hay calificaciones
- Carta Al Autor de La Canción de La BrujaDocumento3 páginasCarta Al Autor de La Canción de La BrujaMoy Aguilar100% (1)
- Actividad 1.3aaguilar Delgado MoisesDocumento5 páginasActividad 1.3aaguilar Delgado MoisesMoy AguilarAún no hay calificaciones
- Actividad 1.2 Aguilar Delgado MoisesDocumento3 páginasActividad 1.2 Aguilar Delgado MoisesMoy AguilarAún no hay calificaciones
- Solubilidad Del Cloruro de BarioDocumento2 páginasSolubilidad Del Cloruro de BarioMoy AguilarAún no hay calificaciones
- Quimica Verde - ADMDocumento6 páginasQuimica Verde - ADMMoy AguilarAún no hay calificaciones
- Quimica Organica - TRabajo FinalDocumento5 páginasQuimica Organica - TRabajo FinalMoy AguilarAún no hay calificaciones
- Milltronics Mfa 4pDocumento35 páginasMilltronics Mfa 4pEduardo Favio Lopez PalominoAún no hay calificaciones
- Practica Tubo VenturiDocumento8 páginasPractica Tubo VenturiLuis CanalesAún no hay calificaciones
- Desarrollo e Innovacion en Ingenieria Vol I 1197161Documento797 páginasDesarrollo e Innovacion en Ingenieria Vol I 1197161Dante Gibran Jasso DiazAún no hay calificaciones
- DNC Et 051Documento17 páginasDNC Et 051rafaelAún no hay calificaciones
- Spa Intex Inst.Documento18 páginasSpa Intex Inst.javierAún no hay calificaciones
- Regulador de TensionDocumento4 páginasRegulador de TensionJonathan Arias MontesAún no hay calificaciones
- Saneamiento Informe Relleno SanitarioDocumento10 páginasSaneamiento Informe Relleno SanitarioErick ChalcoAún no hay calificaciones
- Antecedentes Avances y Desafíos Existentes Del Transporte Internacional de MercancíasDocumento6 páginasAntecedentes Avances y Desafíos Existentes Del Transporte Internacional de MercancíasBrenda VazquezAún no hay calificaciones
- Apuntes Cuantica PDFDocumento43 páginasApuntes Cuantica PDFEnrique Lavilla JulianAún no hay calificaciones
- Eaorozco - 35 - Torque Sobre Una Espira Con Corriente (Dipolo Magnético) en Un Campo Magnético y Energía de Interacción Dipolo-CampoDocumento10 páginasEaorozco - 35 - Torque Sobre Una Espira Con Corriente (Dipolo Magnético) en Un Campo Magnético y Energía de Interacción Dipolo-CampoAntoor 13Aún no hay calificaciones
- Gases de Efecto InvernaderoDocumento11 páginasGases de Efecto InvernaderoJessenia GalvisAún no hay calificaciones
- Libro Sistema SolarDocumento12 páginasLibro Sistema Solarfotocopietas96% (25)
- Circuitos AC en El Estado Senoidal EstableDocumento10 páginasCircuitos AC en El Estado Senoidal EstableGregAún no hay calificaciones
- Luminaria Solar All in OneDocumento4 páginasLuminaria Solar All in OneJuan Miguel Lopez ⑪Aún no hay calificaciones
- Unidad 2 Ciclos TermodinámicosDocumento32 páginasUnidad 2 Ciclos TermodinámicosHarold Fuentes PonceAún no hay calificaciones
- Cámara de CombustiónDocumento15 páginasCámara de CombustiónMirko Alejandro Garcia SilvaAún no hay calificaciones
- Astrid NicolDocumento6 páginasAstrid NicolDaniel ValleAún no hay calificaciones
- Fuerzas Conservativas y Campos GravitatoriosDocumento10 páginasFuerzas Conservativas y Campos GravitatoriosfabricioAún no hay calificaciones
- Capitulo I - Gestión de La Operación de Sistemas de GeneraciónDocumento55 páginasCapitulo I - Gestión de La Operación de Sistemas de GeneraciónElisban H.Aún no hay calificaciones
- Fotosíntesis: ClorofílicaDocumento3 páginasFotosíntesis: ClorofílicaCarol ParedesAún no hay calificaciones
- Informe Karla Salazar Final para Imprimir Listo Ya - Doc YaDocumento42 páginasInforme Karla Salazar Final para Imprimir Listo Ya - Doc Yajessica maria silva filipinoAún no hay calificaciones
- Abrazadera Clip Tipo oDocumento1 páginaAbrazadera Clip Tipo oCARLOS HOMERO TREVIÑO GUADARRAMAAún no hay calificaciones
- Analisis Inmediato de CarbonDocumento6 páginasAnalisis Inmediato de CarbonRosaliaFernandezGutierrezAún no hay calificaciones
- DH de ReaccionDocumento9 páginasDH de ReaccionAgustin VinceAún no hay calificaciones
- Laboratorio 6 de Fisica 3Documento8 páginasLaboratorio 6 de Fisica 3DANIEL ROBINSON HUAMANI RIVEROSAún no hay calificaciones