También podría gustarte
- Bandas Trans PDFDocumento96 páginasBandas Trans PDFYuly XDAún no hay calificaciones
- La Teoría Funcional de La DensidadDocumento4 páginasLa Teoría Funcional de La DensidadDana Alessandra Cardenas VargasAún no hay calificaciones
- Apuntes Quimica SelectividadDocumento33 páginasApuntes Quimica SelectividadrosquilloAún no hay calificaciones
- Mecánica Cuántica Relativista y No Relativista: las dos a la vez: Parte I: Estados estacionariosDe EverandMecánica Cuántica Relativista y No Relativista: las dos a la vez: Parte I: Estados estacionariosAún no hay calificaciones
- 2a - CONSORT - Ensayos Clínicos - Lista de VerificaciónDocumento3 páginas2a - CONSORT - Ensayos Clínicos - Lista de VerificaciónDazaeth Delgado SánchezAún no hay calificaciones
- Plan de Negocio Niñeras A DomicilioDocumento14 páginasPlan de Negocio Niñeras A DomicilioLuisa Elizabeth Rodriguez Parra50% (2)
- La Aproximacion de Hartree - Capitulo de AvanceDocumento16 páginasLa Aproximacion de Hartree - Capitulo de Avancerafael pimentelAún no hay calificaciones
- Rac 8130 - 10Documento2 páginasRac 8130 - 10api-520939289Aún no hay calificaciones
- HerramietascomputacionalesDocumento27 páginasHerramietascomputacionalesDenesis TejedaAún no hay calificaciones
- Clase 2. - Hibridacion ResonanciaDocumento23 páginasClase 2. - Hibridacion ResonanciaEduardo SalinasAún no hay calificaciones
- Enlace Covalente Clase UnoDocumento48 páginasEnlace Covalente Clase UnoAnalia Aiza100% (1)
- Enlace Quimico COVALENTE para ImprimirDocumento52 páginasEnlace Quimico COVALENTE para ImprimirAlejandro RecioAún no hay calificaciones
- Una Aplicacion A Problemas de Fisica de La Materia CondensadaDocumento10 páginasUna Aplicacion A Problemas de Fisica de La Materia CondensadaTaratata Ignacio FelicianoAún no hay calificaciones
- Teoría Del Funcional Densidad DFT (Aplicada A Sistemas Electrónicos)Documento3 páginasTeoría Del Funcional Densidad DFT (Aplicada A Sistemas Electrónicos)Denesis Tejeda100% (1)
- Resueltos Serie 3Documento27 páginasResueltos Serie 3aleinAún no hay calificaciones
- En Laces QuímicosDocumento56 páginasEn Laces QuímicosLesly Janin MercadoAún no hay calificaciones
- Espectros HidrocarburosDocumento7 páginasEspectros Hidrocarburosrgthefenix31Aún no hay calificaciones
- Semana 5 - INO1Documento51 páginasSemana 5 - INO1JOHAN PERCY CHULLUNCUY YUPANQUIAún no hay calificaciones
- 04 Excepciones Lewis y Geometria MolecularDocumento35 páginas04 Excepciones Lewis y Geometria MolecularEve ValdesAún no hay calificaciones
- 3.1. Enlace CovalenteDocumento42 páginas3.1. Enlace CovalenteMARIANA GOMEZ CHAVEZAún no hay calificaciones
- Fisica TeoricaDocumento40 páginasFisica Teoricanapoleon batistaAún no hay calificaciones
- Uniones Químicas - Enlace CovalenteDocumento49 páginasUniones Químicas - Enlace CovalenteJudith SinghAún no hay calificaciones
- Objetivos de La Lección: Variaciones, Que Serán Analizados en Esta LecciónDocumento19 páginasObjetivos de La Lección: Variaciones, Que Serán Analizados en Esta LecciónAtilano jose Cubas aranaAún no hay calificaciones
- Tema 3 - Quimica I - IADocumento65 páginasTema 3 - Quimica I - IARebeca RenedoAún no hay calificaciones
- Universidad Nacional de Tumbes: Numeros Cuanticos Y Trabla PeriodicaDocumento71 páginasUniversidad Nacional de Tumbes: Numeros Cuanticos Y Trabla PeriodicaJohao Olaya espinozaAún no hay calificaciones
- 1 Formulas Lewis TomDocumento14 páginas1 Formulas Lewis TomMax Silverio ValverdeAún no hay calificaciones
- Tarea 10Documento2 páginasTarea 10Berenice MuruagaAún no hay calificaciones
- Moleculas HomonuclearesDocumento19 páginasMoleculas HomonuclearesPecas MonacoAún no hay calificaciones
- HelioDocumento8 páginasHelioWilliam AgudeloAún no hay calificaciones
- HuckelDocumento10 páginasHuckelNico Ariel JaramilloAún no hay calificaciones
- Notas de Teoria de Las Repulsiones Entre Los Pares de Electrones de La Capa de ValenciaDocumento7 páginasNotas de Teoria de Las Repulsiones Entre Los Pares de Electrones de La Capa de ValenciaEgcrisostomoAún no hay calificaciones
- Unidad 5 Parte 3Documento40 páginasUnidad 5 Parte 3ELLEN ORNELLA FIGUEROA CASTROAún no hay calificaciones
- SpectrosDocumento24 páginasSpectrosDaniel Menéndez CrespoAún no hay calificaciones
- FISICA ATOMICA - ApuntesDocumento124 páginasFISICA ATOMICA - ApuntesPablo Carro PortosAún no hay calificaciones
- Tema 4 Bandas-Dinámica 1920Documento65 páginasTema 4 Bandas-Dinámica 1920derfis3Aún no hay calificaciones
- Clase 3 Parte 2Documento57 páginasClase 3 Parte 2Fran Koo VilteAún no hay calificaciones
- Tema 2 Enlace QuimicoDocumento21 páginasTema 2 Enlace QuimicoDavid DeelavillaAún no hay calificaciones
- ESQUEMA-RESUMEN - La Estructura de La MateriaDocumento4 páginasESQUEMA-RESUMEN - La Estructura de La MateriaSilviaLimenAún no hay calificaciones
- Clase 03 - CM10Documento35 páginasClase 03 - CM10AlejandroAún no hay calificaciones
- Cap 1Documento14 páginasCap 1rafael pimentelAún no hay calificaciones
- Taller Sobre Geometria MolecularDocumento3 páginasTaller Sobre Geometria MolecularValentina Pérez100% (1)
- Diapositivas de QuimicaDocumento28 páginasDiapositivas de QuimicaLuis Leonardo Gonzalez MorenoAún no hay calificaciones
- Teoria Quimica 5º AñoDocumento86 páginasTeoria Quimica 5º Añoyuri jesus uzcategui0% (1)
- Tema IV Enlace Químico 2015 Parte IIDocumento69 páginasTema IV Enlace Químico 2015 Parte IIOlmedo Aedo Huyhua AcevedoAún no hay calificaciones
- Experimento de FranckDocumento6 páginasExperimento de FranckMario BubuselaAún no hay calificaciones
- Fisica Moderna Examen Final PDFDocumento4 páginasFisica Moderna Examen Final PDFKATHERINE JESSENIA DELEG PAÑIAún no hay calificaciones
- CementitaDocumento9 páginasCementitaLeonel RuizAún no hay calificaciones
- Estructura y Propiedad de MoléculaDocumento39 páginasEstructura y Propiedad de MoléculaLuisMPortillaBenavidesAún no hay calificaciones
- INFORMEDocumento4 páginasINFORMEKey Yela ʚïɞAún no hay calificaciones
- Consult y Ejercicios Deber 1Documento6 páginasConsult y Ejercicios Deber 1Kevincito PapitoAún no hay calificaciones
- Tema 4Documento27 páginasTema 4javier gagoAún no hay calificaciones
- Seminario 1 - Soluciones PDFDocumento10 páginasSeminario 1 - Soluciones PDFAntonio OrdoñezAún no hay calificaciones
- 07 Conf 4 Enlace Covalente THODocumento59 páginas07 Conf 4 Enlace Covalente THORaúlAún no hay calificaciones
- PEC2Documento5 páginasPEC2lauraAún no hay calificaciones
- HartreeFock 21555Documento17 páginasHartreeFock 21555Pamela YelaAún no hay calificaciones
- Clase 11 2006Documento12 páginasClase 11 2006Abraham Rodriguez HernandezAún no hay calificaciones
- Estructura NuclearDocumento76 páginasEstructura NuclearTesla NoralesAún no hay calificaciones
- Presentación 8 (QUIM 1103)Documento23 páginasPresentación 8 (QUIM 1103)Juan Jose HermosaAún no hay calificaciones
- Tema IIDocumento32 páginasTema IIExequiel RomeoAún no hay calificaciones
- Modelos de Enlace Dr. Noé Zúñiga-VillarrealDocumento98 páginasModelos de Enlace Dr. Noé Zúñiga-VillarrealMishtle TsatsomokaAún no hay calificaciones
- BermudezLuisa-TovarAngie - Informe1Documento12 páginasBermudezLuisa-TovarAngie - Informe1Camila RiverosAún no hay calificaciones
- MasterClass QuantumChemistry UniAndes Week11Documento14 páginasMasterClass QuantumChemistry UniAndes Week11Alejandra GomezAún no hay calificaciones
- Enlace Covalente 2013 BreveDocumento57 páginasEnlace Covalente 2013 BreveLópez Vázquez Carlos YaelAún no hay calificaciones
- Tema 2. Hibridación y EnlacesDocumento78 páginasTema 2. Hibridación y EnlacesAriana HerreraAún no hay calificaciones
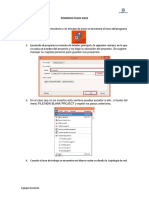
- Primeros Pasos GNS3Documento8 páginasPrimeros Pasos GNS3Abelardo Encinas SilvaAún no hay calificaciones
- Electronic Ticket Orilus SilphiseDocumento3 páginasElectronic Ticket Orilus SilphiseBAIMA RANCAGUAAún no hay calificaciones
- Https WWW - Ejesa.com - Ar Gestion Pwa Factura-Digital InvoiceHeaderToPrint ServiceNumber 236488&InvoiceNumber 49858556Documento1 páginaHttps WWW - Ejesa.com - Ar Gestion Pwa Factura-Digital InvoiceHeaderToPrint ServiceNumber 236488&InvoiceNumber 49858556Lisa BartolettiAún no hay calificaciones
- Brochure Global Solutions 2022Documento12 páginasBrochure Global Solutions 2022samuel carbajal G.Aún no hay calificaciones
- Ejercicio 3 Unidad 3 - Monica CelisDocumento4 páginasEjercicio 3 Unidad 3 - Monica CelisMonica Celis ArizaAún no hay calificaciones
- Material TeóricoDocumento63 páginasMaterial TeóricoMayrin Hernández LeónAún no hay calificaciones
- Educación Física. 2do Año Adtitud y Actitud FisicaDocumento6 páginasEducación Física. 2do Año Adtitud y Actitud FisicaYIRLEY montoyaAún no hay calificaciones
- Sikadur 43 Mortero de ReparaciónDocumento4 páginasSikadur 43 Mortero de ReparaciónVladimir Huamanvilca HuamaniAún no hay calificaciones
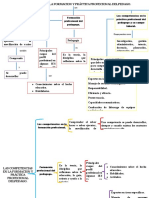
- Competencias Ciudadanas Intento 1Documento6 páginasCompetencias Ciudadanas Intento 1Oscar AgudeloAún no hay calificaciones
- Charlas 25-01Documento20 páginasCharlas 25-01Pablo LucoAún no hay calificaciones
- Campo LaboralDocumento2 páginasCampo LaboralFlor Elena Sanchez CastroAún no hay calificaciones
- Determinación de La Composición Química PDFDocumento114 páginasDeterminación de La Composición Química PDFRudyAún no hay calificaciones
- 3065 03 Suspension de RetencionesDocumento4 páginas3065 03 Suspension de RetencionesPatxy Lopez ChacaliazaAún no hay calificaciones
- Comunicado Joel JaraDocumento2 páginasComunicado Joel JaraTransgenicos CultivaAún no hay calificaciones
- LOMBRIFILTRODocumento51 páginasLOMBRIFILTROMariel Oblitas100% (1)
- Comparto - Gato - Minnie .En - Es - ContigoDocumento11 páginasComparto - Gato - Minnie .En - Es - Contigofercho-javierAún no hay calificaciones
- Aromas FenpalDocumento9 páginasAromas FenpalFran MorenoAún no hay calificaciones
- Reglamento para Grupo - Interes-2018Documento7 páginasReglamento para Grupo - Interes-2018Cesario Cruz100% (1)
- 31 Hta NeumaticaDocumento20 páginas31 Hta Neumaticagabriell_189Aún no hay calificaciones
- Acoplamientos FlexiblesDocumento22 páginasAcoplamientos FlexiblesClaudia Mesias Ochoa100% (1)
- Taller No. 1 Matriz de Identificación de Documentación Requerida para Los SG HSEQDocumento5 páginasTaller No. 1 Matriz de Identificación de Documentación Requerida para Los SG HSEQMary cruz SegoviaAún no hay calificaciones
- Señora: Elin Vasquez Paternina Ciudad Referencia: Radicado SSPD No. 20215291147112 Del 25 de Mayo Expediente No. 2021820380100095EDocumento2 páginasSeñora: Elin Vasquez Paternina Ciudad Referencia: Radicado SSPD No. 20215291147112 Del 25 de Mayo Expediente No. 2021820380100095EElín Vásquez PaterninaAún no hay calificaciones
- Manual Smart Led TV Hyundai Modelos Hyled 32HDSG Hyled 43FHDS PDFDocumento28 páginasManual Smart Led TV Hyundai Modelos Hyled 32HDSG Hyled 43FHDS PDFSoportes SolucionesAún no hay calificaciones
- SolarisDocumento50 páginasSolarisJulian VPAún no hay calificaciones
- Esta Di SticaDocumento10 páginasEsta Di SticaMarce MosxqueraAún no hay calificaciones
- 4° Portafolio C.T.S. 1Documento16 páginas4° Portafolio C.T.S. 1Julio Santiago Cotrina100% (1)