También podría gustarte
- Validacion de Procesos en La Industria FarmaceuticaDocumento37 páginasValidacion de Procesos en La Industria FarmaceuticaASEGURAMIENTO DE LA CALIDADAún no hay calificaciones
- Taller 4. Plan de CalidadDocumento2 páginasTaller 4. Plan de CalidadTarcila CadizAún no hay calificaciones
- PQP: Proceso de calificación de partesDocumento61 páginasPQP: Proceso de calificación de partesluisAún no hay calificaciones
- Despliegue de La Función de Calidad IIDocumento70 páginasDespliegue de La Función de Calidad IIFernandoAún no hay calificaciones
- IV Validación de Procesos PDFDocumento37 páginasIV Validación de Procesos PDFmariAún no hay calificaciones
- Proc de MCP Cil HydDocumento1 páginaProc de MCP Cil HydSaul Huaman SanchezAún no hay calificaciones
- Anexo 7 Peig-Sgc-M-F-0 Plan de Inspeccion y EnsayoDocumento16 páginasAnexo 7 Peig-Sgc-M-F-0 Plan de Inspeccion y EnsayoNeomar Velasquez100% (1)
- FOR-LAB-006 - Matriz - LV 17025 - Crit - Pol - Ley - SECCION - C - EA - 06AIDADocumento7 páginasFOR-LAB-006 - Matriz - LV 17025 - Crit - Pol - Ley - SECCION - C - EA - 06AIDAAngel SevillaAún no hay calificaciones
- IndicepipingDocumento1 páginaIndicepipingjuniuni18Aún no hay calificaciones
- FIAC-157 Control de Cambios Movimiento Maquina Automatizada Broom Rev 4Documento1 páginaFIAC-157 Control de Cambios Movimiento Maquina Automatizada Broom Rev 4luis.rangelAún no hay calificaciones
- 01 Ppi Topografia v1Documento2 páginas01 Ppi Topografia v1Wara VelasquezAún no hay calificaciones
- RG04-IO3501 Plan de CalidadDocumento7 páginasRG04-IO3501 Plan de CalidadKirita StAún no hay calificaciones
- Bitácora Curso Iso 22301 Sesión 2Documento6 páginasBitácora Curso Iso 22301 Sesión 2Angie Evangelista MautinoAún no hay calificaciones
- Sistema de La Gestion de La Calidad (Paradas de Planta)Documento2 páginasSistema de La Gestion de La Calidad (Paradas de Planta)Ing. Jhon RomeroAún no hay calificaciones
- Archivos Adjuntos OutlookArticulo de Commissioning & ValidaciónDocumento22 páginasArchivos Adjuntos OutlookArticulo de Commissioning & ValidaciónNadia WilsonAún no hay calificaciones
- Proceso certificación IMSSDocumento7 páginasProceso certificación IMSSMons PoketAún no hay calificaciones
- Plan de Trabajo Estudiante (Quimica) Ucv MJDocumento4 páginasPlan de Trabajo Estudiante (Quimica) Ucv MJRonny PortilloAún no hay calificaciones
- Ficha de Revisión - Plan y Programa de Auditoría de ElectroDocumento15 páginasFicha de Revisión - Plan y Programa de Auditoría de ElectroalexAún no hay calificaciones
- Matriz Evaluacion de SoftwareDocumento9 páginasMatriz Evaluacion de SoftwareFatima PavonAún no hay calificaciones
- Control Operacional - ICONTECDocumento31 páginasControl Operacional - ICONTECEdinson Pacheco MoralesAún no hay calificaciones
- Clase 7Documento17 páginasClase 7Diego YarlequeAún no hay calificaciones
- Ficha de Revisión - Plan y Programa de AuditoríaDocumento14 páginasFicha de Revisión - Plan y Programa de AuditoríaBielMamaniYapuchuraAún no hay calificaciones
- Sistemas Criticos Validacion y Verificacion Continua Del ProcesoDocumento105 páginasSistemas Criticos Validacion y Verificacion Continua Del ProcesoIlze Belen Medina MaderaAún no hay calificaciones
- Definición, planeación y ejecución de proyectosDocumento1 páginaDefinición, planeación y ejecución de proyectosjasg001Aún no hay calificaciones
- Definición, planeación y ejecución de proyectosDocumento1 páginaDefinición, planeación y ejecución de proyectosjasg001Aún no hay calificaciones
- Manual de Calidad Adoquines S.A.CDocumento43 páginasManual de Calidad Adoquines S.A.CBibiana Paola Lezama RuizAún no hay calificaciones
- Caso UsoDocumento1 páginaCaso UsoNestorAún no hay calificaciones
- Flujo de ProyectosDocumento2 páginasFlujo de ProyectosCHAKA TRAKAAún no hay calificaciones
- Calidad Six SigmaDocumento1 páginaCalidad Six Sigmaca4707394Aún no hay calificaciones
- Entrenamiento AMT - Planeacion MDCDocumento126 páginasEntrenamiento AMT - Planeacion MDCIsaias OrsiniAún no hay calificaciones
- Validación analítica de métodos en laboratorioDocumento11 páginasValidación analítica de métodos en laboratorioHugo Huaman MuñozAún no hay calificaciones
- Core Tools PresentaciónDocumento151 páginasCore Tools Presentaciónaugusto5891Aún no hay calificaciones
- Gestión Cal Ind FarmDocumento32 páginasGestión Cal Ind FarmmaraperezlindoAún no hay calificaciones
- RG04-IO3501 Plan de CalidadDocumento5 páginasRG04-IO3501 Plan de Calidadmaria isabelAún no hay calificaciones
- Change ManagementDocumento1 páginaChange ManagementErika Alejandra Mecias AndradeAún no hay calificaciones
- PIE-SICOLL-2024-01, Rev.0 Pba. Hidrostática Collahuasi Línea de 7 PulgDocumento4 páginasPIE-SICOLL-2024-01, Rev.0 Pba. Hidrostática Collahuasi Línea de 7 PulgRodrigoArayaAún no hay calificaciones
- 1 Matriz de Riesgos y Oportunidades Del SGDocumento2 páginas1 Matriz de Riesgos y Oportunidades Del SGDiana Marisell Carrera LLaxaAún no hay calificaciones
- Ficha Tecnica de Indicadores CualitativosdocxDocumento2 páginasFicha Tecnica de Indicadores Cualitativosdocxestefany9507100% (1)
- Matriz de Consistencia Empresa ApropalDocumento3 páginasMatriz de Consistencia Empresa ApropalLuis Chavez reyesAún no hay calificaciones
- Gestion Por ProcesosDocumento26 páginasGestion Por Procesosjmontalvoa2Aún no hay calificaciones
- CRONOGRAMA AUDITORIA 2016Documento5 páginasCRONOGRAMA AUDITORIA 2016nelson omar restrepo loaizaAún no hay calificaciones
- Lo-Fr-06 Seleccion de ProveedoresDocumento1 páginaLo-Fr-06 Seleccion de Proveedoresjj galeanoAún no hay calificaciones
- 13) Plan de Control Rev 3.0Documento18 páginas13) Plan de Control Rev 3.0Dipol AutomotiveAún no hay calificaciones
- Motores 2Documento30 páginasMotores 2Alex TacuriAún no hay calificaciones
- Herramientas de CalidadDocumento27 páginasHerramientas de CalidadMaria del Carmen Salazar EspinozaAún no hay calificaciones
- GR-FO-05 Selección y Aprobación Inicial de ProveedoresDocumento3 páginasGR-FO-05 Selección y Aprobación Inicial de ProveedoresehernandezmesaAún no hay calificaciones
- Gestión de La Calidad en Los NegociosDocumento3 páginasGestión de La Calidad en Los NegociosChonchito NoriegaAún no hay calificaciones
- Implementación SGC Diagrama FlujoDocumento3 páginasImplementación SGC Diagrama FlujoChonchito NoriegaAún no hay calificaciones
- Gestion de Calidad Tarea Semana 2Documento5 páginasGestion de Calidad Tarea Semana 2Rodrigo araya baltierraAún no hay calificaciones
- Flujograma GeneralDocumento1 páginaFlujograma Generaljohoran.fierrocAún no hay calificaciones
- Calibración de instrumentosDocumento5 páginasCalibración de instrumentosjuan olarteAún no hay calificaciones
- Estado de equipos de medición Planta Cobre ALCALDEDocumento4 páginasEstado de equipos de medición Planta Cobre ALCALDEkelvynAún no hay calificaciones
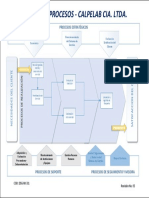
- DOC-SGC-04 Mapa de Procesos SicepDocumento2 páginasDOC-SGC-04 Mapa de Procesos SicepMaria Elena SarmientoAún no hay calificaciones
- S10-Fase Controlar Six SigmaDocumento18 páginasS10-Fase Controlar Six SigmaJorge Rojas GeldresAún no hay calificaciones
- Mapa de ProcesosDocumento1 páginaMapa de ProcesosChris SeguraAún no hay calificaciones
- Habilitación y Montaje de TuberíaDocumento1 páginaHabilitación y Montaje de TuberíaLuiz Aburto AguirreAún no hay calificaciones
- Diagrama lógico mantenimiento sistema producciónDocumento1 páginaDiagrama lógico mantenimiento sistema producciónJorge Enrique Tello Estrada100% (1)
- 3059-P3-PPI-EST-023 Montaje de Estructuras MetálicasDocumento1 página3059-P3-PPI-EST-023 Montaje de Estructuras Metálicaseddy tincoAún no hay calificaciones
- PO-SGC-00-015 Rev 0 Control de Equipos de Inspección y EnsayoDocumento7 páginasPO-SGC-00-015 Rev 0 Control de Equipos de Inspección y EnsayoarieljudaAún no hay calificaciones
- Práctica - Procedimientos de Operación Estandarizados - 2023Documento29 páginasPráctica - Procedimientos de Operación Estandarizados - 2023KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- P7-Control de Calidad Por AtributosDocumento9 páginasP7-Control de Calidad Por AtributosKATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- Práctica 4Documento5 páginasPráctica 4KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- Práctica 4Documento5 páginasPráctica 4KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- Buenas Prácticas de Manufactura - 2023 - IDocumento99 páginasBuenas Prácticas de Manufactura - 2023 - IKATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- P11 - Calibración de Material Volumétrico.Documento16 páginasP11 - Calibración de Material Volumétrico.KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- Sesion 09 Validaciones Tecnologia 2023Documento92 páginasSesion 09 Validaciones Tecnologia 2023KATERIN THALIA RONDO TOLENTINOAún no hay calificaciones
- ACTIVIDADES Quimica General Proyecto 2.0Documento13 páginasACTIVIDADES Quimica General Proyecto 2.0Cristian MoraAún no hay calificaciones
- Aeronavegabilidad, Confiabilidad y ElegibilidadDocumento31 páginasAeronavegabilidad, Confiabilidad y ElegibilidadJORGE ENRIQUE CHAPA TUME100% (1)
- Impermeabilizacion DIMARTIDocumento32 páginasImpermeabilizacion DIMARTIJuan Jose AyalaAún no hay calificaciones
- Ficha de Actividades 3 Analisis DimensionalDocumento4 páginasFicha de Actividades 3 Analisis Dimensionalyangos yangos100% (1)
- Propagacion de ErroresDocumento10 páginasPropagacion de ErroresLiliana Timana InsandaraAún no hay calificaciones
- 144 Prob Estadi.Documento6 páginas144 Prob Estadi.arielAún no hay calificaciones
- 3 - Cortante en Los SuelosDocumento57 páginas3 - Cortante en Los SuelosLEIDY VIVIANA GUTIERREZ GALINDOAún no hay calificaciones
- Iperc - Montaje Puente Perene Consorcio CapachariDocumento22 páginasIperc - Montaje Puente Perene Consorcio CapachariSSoma Prevencionista80% (5)
- Informe #002 de Compatibilidad de Expediente TecnicoDocumento8 páginasInforme #002 de Compatibilidad de Expediente TecnicoDavidVegaAún no hay calificaciones
- Report V 1235Documento1 páginaReport V 1235AlixiAún no hay calificaciones
- Leto - The Real Master Reel PDFDocumento75 páginasLeto - The Real Master Reel PDFDavid100% (1)
- Catalogo Tecnico Siding PLYCENDocumento8 páginasCatalogo Tecnico Siding PLYCENMario MendezAún no hay calificaciones
- Formato Voz Del ClienteDocumento14 páginasFormato Voz Del ClienteLau ZambranoAún no hay calificaciones
- Practica 01 SOTO HUAMÁNDocumento14 páginasPractica 01 SOTO HUAMÁNNorman Morales MontañoAún no hay calificaciones
- Geotexil No TejidoDocumento4 páginasGeotexil No TejidorossmerypacocunyaAún no hay calificaciones
- Numeracion 4to AnoDocumento2 páginasNumeracion 4to AnoGean Pierre C. BernedoAún no hay calificaciones
- Crocodile electricidad prácticasDocumento15 páginasCrocodile electricidad prácticasFranklin Altamirano PadillaAún no hay calificaciones
- 4° CCPC ELECTRICIDAD Guia2Documento6 páginas4° CCPC ELECTRICIDAD Guia2Javier SuazoAún no hay calificaciones
- Tecnologia Mecanismos 5 PDFDocumento22 páginasTecnologia Mecanismos 5 PDFJOSE LUIS LOPEZ RECHEAún no hay calificaciones
- Repuestos SAME de La Mejor CalidadDocumento2 páginasRepuestos SAME de La Mejor CalidadFederico EnphasAún no hay calificaciones
- Taller Potencia Fluida - Bombas y MotoresDocumento11 páginasTaller Potencia Fluida - Bombas y MotoresJulianManriqueAún no hay calificaciones
- Fisica 2 Tema1Documento7 páginasFisica 2 Tema1Khrriz CastilloAún no hay calificaciones
- Pistola SemiautomaticaDocumento24 páginasPistola SemiautomaticajulioAún no hay calificaciones
- Investigacion de CarreterasDocumento4 páginasInvestigacion de CarreterasNepta Lope GomezzAún no hay calificaciones
- Foro 2Documento5 páginasForo 2Carlos Méndez100% (1)
- 3.materiales Amorfos y CristalinosDocumento40 páginas3.materiales Amorfos y CristalinosPabloAún no hay calificaciones
- Informe PoechosDocumento14 páginasInforme PoechosBoulangger Tume Arturo0% (1)
- Ablandador de AguaDocumento2 páginasAblandador de AguaJean Pierre Pinedo BardalesAún no hay calificaciones
- Evaluación Densidad Aparente NCH 1116 of 77Documento2 páginasEvaluación Densidad Aparente NCH 1116 of 77JOSEAún no hay calificaciones
- Practica 1 Maquinas Electricas IIDocumento4 páginasPractica 1 Maquinas Electricas IICamilo ArdilaAún no hay calificaciones
- Fundamentos de producción y gestión de proyectos audiovisualesDe EverandFundamentos de producción y gestión de proyectos audiovisualesCalificación: 5 de 5 estrellas5/5 (2)
- Calidad y servicio. Concepto y herramientasDe EverandCalidad y servicio. Concepto y herramientasAún no hay calificaciones
- Mejoramiento de la calidad. Un enfoque a serviciosDe EverandMejoramiento de la calidad. Un enfoque a serviciosCalificación: 4.5 de 5 estrellas4.5/5 (7)
- Proyecte su jardín como un profesionalDe EverandProyecte su jardín como un profesionalCalificación: 5 de 5 estrellas5/5 (1)
- Apuntes de diseño de interiores: Principios básicos de escalas, espacios, colores y másDe EverandApuntes de diseño de interiores: Principios básicos de escalas, espacios, colores y másCalificación: 4.5 de 5 estrellas4.5/5 (20)
- Marketing de contenidos. Guía prácticaDe EverandMarketing de contenidos. Guía prácticaCalificación: 4.5 de 5 estrellas4.5/5 (15)
- Manual NSCA: Fundamentos del entrenamiento personalDe EverandManual NSCA: Fundamentos del entrenamiento personalCalificación: 5 de 5 estrellas5/5 (8)
- Diseño urbano: Teoría y Método. Tercera edición actualizadaDe EverandDiseño urbano: Teoría y Método. Tercera edición actualizadaAún no hay calificaciones
- Fundamentos del diseño y la construcción con maderaDe EverandFundamentos del diseño y la construcción con maderaCalificación: 3 de 5 estrellas3/5 (5)
- Cambio climático: Lecciones de y para ciudades de América LatinaDe EverandCambio climático: Lecciones de y para ciudades de América LatinaCalificación: 4 de 5 estrellas4/5 (2)
- Tendencias de la investigación en ingeniería ambientalDe EverandTendencias de la investigación en ingeniería ambientalCalificación: 5 de 5 estrellas5/5 (1)
- Gestión Administrativa y Comercial en RestauraciónDe EverandGestión Administrativa y Comercial en RestauraciónAún no hay calificaciones
- Ciclo de vida de los productos. Diseño y análisis para la innovación sostenibleDe EverandCiclo de vida de los productos. Diseño y análisis para la innovación sostenibleCalificación: 3 de 5 estrellas3/5 (2)
- Calidad y servicio: Conceptos y herramientasDe EverandCalidad y servicio: Conceptos y herramientasCalificación: 5 de 5 estrellas5/5 (2)
- Revit MEP 2018 Curso Práctico: Diseño asistido por ordenador (CAD)De EverandRevit MEP 2018 Curso Práctico: Diseño asistido por ordenador (CAD)Calificación: 2.5 de 5 estrellas2.5/5 (6)
- Tipologías arquitectónicas coloniales y republicanas: Afinidades y oposiciones. Cartagena de Indias, Turbaco, ArjonaDe EverandTipologías arquitectónicas coloniales y republicanas: Afinidades y oposiciones. Cartagena de Indias, Turbaco, ArjonaCalificación: 4 de 5 estrellas4/5 (3)
- Hidroponía. Cultivo sin tierraDe EverandHidroponía. Cultivo sin tierraCalificación: 4.5 de 5 estrellas4.5/5 (15)
- El método Seis Sigma: Mejore los resultados de su negocioDe EverandEl método Seis Sigma: Mejore los resultados de su negocioCalificación: 4 de 5 estrellas4/5 (24)
- Técnicas de modelos en el proceso de creación y diseño de productosDe EverandTécnicas de modelos en el proceso de creación y diseño de productosCalificación: 4.5 de 5 estrellas4.5/5 (2)
- Arquitectura alternativa sostenibleDe EverandArquitectura alternativa sostenibleCalificación: 5 de 5 estrellas5/5 (1)