También podría gustarte
- Psicometría. Principios básicos y protocolos experimentales diversosDe EverandPsicometría. Principios básicos y protocolos experimentales diversosAún no hay calificaciones
- Conceptos de Fugacidad y ActividadDocumento10 páginasConceptos de Fugacidad y ActividadMauro SuZef67% (3)
- Ecuación de Estado de RedlichDocumento13 páginasEcuación de Estado de RedlichAlis Carbajal PinedaAún no hay calificaciones
- Actividad y Coeficiente de ActividadDocumento24 páginasActividad y Coeficiente de ActividadTania Flores100% (2)
- Coeficiente de ActividadDocumento2 páginasCoeficiente de ActividadMontse CruzAún no hay calificaciones
- Practica 6. Sintesis de Alquenos (Etileno)Documento10 páginasPractica 6. Sintesis de Alquenos (Etileno)Javier AyalaAún no hay calificaciones
- La Teroria de Las Velocidades AbsolutasDocumento7 páginasLa Teroria de Las Velocidades AbsolutasIsai Keoma Chirinos DiazAún no hay calificaciones
- Actividad y Coeficiente de ActividadDocumento4 páginasActividad y Coeficiente de ActividadCrishtian Perez MenesesAún no hay calificaciones
- Tarea Preexamen de Fisicoquímica IDocumento15 páginasTarea Preexamen de Fisicoquímica IAlexis Cisneros100% (1)
- FISICOQUIMICA2Documento14 páginasFISICOQUIMICA2Jesus Alexis Cervantes RamirezAún no hay calificaciones
- Propiedades CríticasDocumento5 páginasPropiedades CríticasPaola Carpio75% (4)
- Orta Pérez - PL2 y 3pdfDocumento35 páginasOrta Pérez - PL2 y 3pdfOsmyy OrtaAún no hay calificaciones
- 1.5. FisicoquimicaDocumento8 páginas1.5. FisicoquimicaMith EcheloonAún no hay calificaciones
- Modelos de SolucionDocumento5 páginasModelos de SolucionDaniel Gonzales ZayasAún no hay calificaciones
- Cuestiones para Discutir Capitulo 2Documento3 páginasCuestiones para Discutir Capitulo 2Fernando Cordova PardoAún no hay calificaciones
- 2.2 Potencial QuimicoDocumento3 páginas2.2 Potencial QuimicoJulianNacimAsAún no hay calificaciones
- Catálisis Homogénea Por Compuestos de CoordinaciónDocumento32 páginasCatálisis Homogénea Por Compuestos de CoordinaciónJohan Delgado100% (1)
- Sistemas No IdealesDocumento36 páginasSistemas No IdealesArturo CollazosAún no hay calificaciones
- Ley de Fick2.01Documento21 páginasLey de Fick2.01Kristian Moreta0% (1)
- Guía de Problemas Equilibrio Líquido VaporDocumento4 páginasGuía de Problemas Equilibrio Líquido VaporGerson SilvaAún no hay calificaciones
- Problemas Resueltos Del Tema 1Documento9 páginasProblemas Resueltos Del Tema 1Ángela Muñoz Chicharro50% (2)
- Propiedades Coligativas Ec. MargulesDocumento57 páginasPropiedades Coligativas Ec. MargulesricardoAún no hay calificaciones
- Gonzalez Hernandez t1Documento9 páginasGonzalez Hernandez t1Veneno Alan Joqsan100% (2)
- Laboratorio 2 ELECTROGRAVIMETRIADocumento12 páginasLaboratorio 2 ELECTROGRAVIMETRIAJuan Pablo Squella100% (1)
- Cap 10Documento25 páginasCap 10Jessy RamirezAún no hay calificaciones
- Mezcla IdealDocumento3 páginasMezcla IdealMiguel Angel Paredes HernándezAún no hay calificaciones
- UNIDAD 5 FLUJO DE EFECTIVO Docx 1Documento25 páginasUNIDAD 5 FLUJO DE EFECTIVO Docx 1Thane Botticelli100% (2)
- Balances MolaresDocumento7 páginasBalances MolaresAlejandro MerkAún no hay calificaciones
- Potenciales Termodinámicos y Criterios de Espontaneidad y EquilibrioDocumento15 páginasPotenciales Termodinámicos y Criterios de Espontaneidad y EquilibrioDaniel Melo100% (1)
- AzeotropiaDocumento25 páginasAzeotropiaRicardo KobainAún no hay calificaciones
- Serie 3 - Equilibrio de Fases - ResueltosDocumento5 páginasSerie 3 - Equilibrio de Fases - ResueltosDidier DetchemendyAún no hay calificaciones
- Formulario Basico Fenómenos de Transporte PDFDocumento2 páginasFormulario Basico Fenómenos de Transporte PDFCristian SinghAún no hay calificaciones
- Coeficiente de Actividad InformeDocumento5 páginasCoeficiente de Actividad InformeJesús Cristhian ChipanaAún no hay calificaciones
- Equilibrio Liquido-Liquido PDFDocumento12 páginasEquilibrio Liquido-Liquido PDFAle Jaime0% (1)
- Aplicaciones de Las Propiedades ColigativasDocumento14 páginasAplicaciones de Las Propiedades ColigativasAlbert V. EspinozaAún no hay calificaciones
- Difusión Molecular en Gases (1.4)Documento5 páginasDifusión Molecular en Gases (1.4)Arlen MamaniAún no hay calificaciones
- Practica 2 SemibatchDocumento26 páginasPractica 2 SemibatchJose Manuel Saldaña Ortiz100% (1)
- Metodo Del Calculo de Transferencia de Calor Por Metodo Ntu y DMLTDocumento10 páginasMetodo Del Calculo de Transferencia de Calor Por Metodo Ntu y DMLTMiguel CastroAún no hay calificaciones
- Actividad y Coef de Act FISICOQUIMICA IDocumento15 páginasActividad y Coef de Act FISICOQUIMICA IOswaldo Rodriguez50% (2)
- Contradifusión Equimolar Parte 3Documento6 páginasContradifusión Equimolar Parte 3José GuacollanteAún no hay calificaciones
- Cuestiones para Discutir Capítulo 8Documento3 páginasCuestiones para Discutir Capítulo 8Isaac Pineda Carbajal100% (2)
- Propiedades Termodinamicas de Las SolucionesDocumento11 páginasPropiedades Termodinamicas de Las SolucionesUlisesYazielSosaLopezAún no hay calificaciones
- Condensadores de Vapor MezcladosDocumento3 páginasCondensadores de Vapor MezcladosAnaGomezAún no hay calificaciones
- 06 Problemas Unidad 2Documento11 páginas06 Problemas Unidad 2Maya MendozaAún no hay calificaciones
- Por Ciento de Conversión, Selectividad y Rendimiento de Una ReacciónDocumento12 páginasPor Ciento de Conversión, Selectividad y Rendimiento de Una ReacciónAlexis R AC50% (2)
- Práctica Volumen Molar ParcialDocumento17 páginasPráctica Volumen Molar ParcialMelissa Salgado100% (1)
- Método de Las Velocidades InicialesDocumento5 páginasMétodo de Las Velocidades InicialesFitoFuckAún no hay calificaciones
- Potencial Químico de Un Gas Ideal en Una MezclaDocumento2 páginasPotencial Químico de Un Gas Ideal en Una MezclaChicho XixiAún no hay calificaciones
- Cuestionario 9 Fenomenos de TransporteDocumento6 páginasCuestionario 9 Fenomenos de TransporteOscarMarin100% (1)
- 09-Transferencia de Masa Conveccion Natural y Forzada 2015 Rev2 PDFDocumento7 páginas09-Transferencia de Masa Conveccion Natural y Forzada 2015 Rev2 PDFRolando RivasAún no hay calificaciones
- Ejercicios para Resolver Diseño ReactorDocumento1 páginaEjercicios para Resolver Diseño Reactorbeto nuñezAún no hay calificaciones
- La FugacidadDocumento5 páginasLa FugacidadMaria Alejandra Narvaez Izaguirre83% (6)
- Ley de Henry y OtrosDocumento6 páginasLey de Henry y OtrosCarlos CcqAún no hay calificaciones
- Las Disoluciones Se Forman Cuando Las Fuerzas de Atracción Entre Las PartículasDocumento2 páginasLas Disoluciones Se Forman Cuando Las Fuerzas de Atracción Entre Las PartículasDiego MuñozAún no hay calificaciones
- Ley de HenrryDocumento7 páginasLey de HenrryJhon ChavezAún no hay calificaciones
- Equilibrio Liquido Gas-FisicoquimicaDocumento34 páginasEquilibrio Liquido Gas-FisicoquimicaYurika Toledo100% (2)
- Disoluciones IdealesDocumento9 páginasDisoluciones IdealesSteven Hicks100% (2)
- Teoría Del Gas Real: La Relación Exacta Para Gases RealesDe EverandTeoría Del Gas Real: La Relación Exacta Para Gases RealesAún no hay calificaciones
- Parra Avila Omar DiegoDocumento1 páginaParra Avila Omar DiegoOmar Diego ParraAún no hay calificaciones
- Cambios en El Rol de La Mujer Desde 1945Documento16 páginasCambios en El Rol de La Mujer Desde 1945Omar Diego ParraAún no hay calificaciones
- NotasDocumento2 páginasNotasOmar Diego ParraAún no hay calificaciones
- COMPENDIO 03 - Gestion Ambiental PDFDocumento316 páginasCOMPENDIO 03 - Gestion Ambiental PDFOmar Diego ParraAún no hay calificaciones
- Diapositivas Transferencia de Calor 2018-IDocumento60 páginasDiapositivas Transferencia de Calor 2018-IOmar Diego ParraAún no hay calificaciones
- Proceso de La CervezaDocumento12 páginasProceso de La CervezaOmar Diego ParraAún no hay calificaciones
- Dialnet TrazosDeOtraComunicacionEnAmericaLatina 537810Documento128 páginasDialnet TrazosDeOtraComunicacionEnAmericaLatina 537810Doyler LastreAún no hay calificaciones
- 03 UANCV EXOSICION Formulas Polinomicas 20 12 2019 PDFDocumento38 páginas03 UANCV EXOSICION Formulas Polinomicas 20 12 2019 PDFAlejandro HCAún no hay calificaciones
- 1 - Carpintería de Interior y de ExteriorDocumento23 páginas1 - Carpintería de Interior y de ExteriorausiasmarchAún no hay calificaciones
- Fisica Calor y Temperatura 01Documento7 páginasFisica Calor y Temperatura 01luisfrojas64Aún no hay calificaciones
- Reactores en SuspensiónDocumento19 páginasReactores en SuspensiónJess Vigoci50% (2)
- Ingenieria Quimica en BoliviaDocumento24 páginasIngenieria Quimica en BoliviaEduardo Hilari0% (1)
- Presupuesto Casa 4 (Con Adicion)Documento1 páginaPresupuesto Casa 4 (Con Adicion)DUBIER ALEJANDRO ARIAS GOMEZAún no hay calificaciones
- Acero 4041Documento6 páginasAcero 4041Winson Marza VeraAún no hay calificaciones
- Cobre y AleacionesDocumento32 páginasCobre y AleacionesRicardo RomeroAún no hay calificaciones
- Microsoft Word - Generalidades Acero para PlasticoDocumento5 páginasMicrosoft Word - Generalidades Acero para PlasticoAnonymous UQiuYdAún no hay calificaciones
- Catalogo Lubricantes Industria tcm7-586754 tcm13-37189Documento57 páginasCatalogo Lubricantes Industria tcm7-586754 tcm13-37189Jhonmar JhonmarAún no hay calificaciones
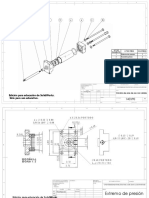
- Ensamble de Piston Neumatico.Documento12 páginasEnsamble de Piston Neumatico.JaimeAún no hay calificaciones
- ElectroquimicaDocumento3 páginasElectroquimicaEdison MataAún no hay calificaciones
- Resumen Ejecutivo-Calidad Del AguaDocumento17 páginasResumen Ejecutivo-Calidad Del AguaJose Felix Diaz TapiaAún no hay calificaciones
- Actividad #2 (Química Inorganíca)Documento11 páginasActividad #2 (Química Inorganíca)Yennifer ARCINIEGASAún no hay calificaciones
- Semana5 Resistencia MaterialesDocumento9 páginasSemana5 Resistencia Materialeskatherine andrea vásquez valdiviaAún no hay calificaciones
- Es Kendall SHP Diesel Engine OilDocumento3 páginasEs Kendall SHP Diesel Engine OilHubertt Chacon AntonioAún no hay calificaciones
- Química Examen Semana 2Documento3 páginasQuímica Examen Semana 2David CruzAún no hay calificaciones
- EQ63 - Aire Como Materia Prima PDFDocumento19 páginasEQ63 - Aire Como Materia Prima PDFandreacarcedo100% (1)
- Informe 6Documento4 páginasInforme 6Carol RamirezAún no hay calificaciones
- Proceso de Elaboracion Del Azucar Rubia 2013Documento4 páginasProceso de Elaboracion Del Azucar Rubia 2013jose ruiz quiroz100% (1)
- Espci 00005 (Eett) Anclajes PostensadosDocumento24 páginasEspci 00005 (Eett) Anclajes Postensadosmatias.josue1996Aún no hay calificaciones
- ELASTICIDADDocumento2 páginasELASTICIDADandres osunaAún no hay calificaciones
- Diplomado Gestión de Residuos SólidosDocumento7 páginasDiplomado Gestión de Residuos SólidosJuan PabloAún no hay calificaciones
- Tesis ResúmenDocumento43 páginasTesis ResúmenPEDRO RAMÓN PATAZCA ROJAS50% (2)
- Cuestionario 1 JahsDocumento19 páginasCuestionario 1 JahsLeonardo Cardona ramirezAún no hay calificaciones
- Aleaciones MetalicasDocumento39 páginasAleaciones MetalicasAndres JimenezAún no hay calificaciones
- Esencia de HierbabuenaDocumento8 páginasEsencia de HierbabuenaHelen H RamAún no hay calificaciones
- Grupo No.3-Seccion XiiiDocumento15 páginasGrupo No.3-Seccion XiiiEsthefanny MirandaAún no hay calificaciones
- MolaridadDocumento3 páginasMolaridadMilton Rolando Ac MaczAún no hay calificaciones