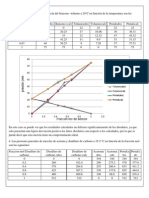
También podría gustarte
- Nuevos usos para viejos medicamentosDe EverandNuevos usos para viejos medicamentosAún no hay calificaciones
- Resultados Fuera de EspecificaciónDocumento3 páginasResultados Fuera de EspecificaciónDalid Chama Coria50% (2)
- Farmacometría:Curvas dosis-respuesta de tipo gradual. Volumen 1De EverandFarmacometría:Curvas dosis-respuesta de tipo gradual. Volumen 1Aún no hay calificaciones
- Estudio de BioequivalenciaDocumento14 páginasEstudio de BioequivalenciaVladimir Condori Melgar0% (1)
- Reporte ReologiaDocumento5 páginasReporte Reologiacnthpl100% (1)
- Manejo de Materiales Empresa AlimenticiaDocumento2 páginasManejo de Materiales Empresa Alimenticiaangelesmtz69927Aún no hay calificaciones
- 0299 Uniformidad de DosisDocumento8 páginas0299 Uniformidad de DosisEdwinAún no hay calificaciones
- PROTOCOLO TABLETAS DE ACICLOVIR DE 200mgDocumento9 páginasPROTOCOLO TABLETAS DE ACICLOVIR DE 200mgJose OlveraAún no hay calificaciones
- Perfil de Discolución en Ácido AcetilsalicílicoDocumento10 páginasPerfil de Discolución en Ácido AcetilsalicílicoRafael Galan100% (1)
- Control de Calidad en El LaboratorioDocumento15 páginasControl de Calidad en El LaboratorioCarlos Gonzalez Callejas0% (1)
- Nom 078 Ssa1 1994Documento8 páginasNom 078 Ssa1 1994Eduardo A Galeana SánchezAún no hay calificaciones
- Tarea Sobre CRIMINALISTICA EN MEXICO de Salazar Muñoz Samantha JazmínDocumento15 páginasTarea Sobre CRIMINALISTICA EN MEXICO de Salazar Muñoz Samantha JazmínMiguel Arturo Herrera Sanchez100% (1)
- Análisis y Control de Medicamentos y Afines IIDocumento11 páginasAnálisis y Control de Medicamentos y Afines IINedyVictoriaBeizagaOliveraAún no hay calificaciones
- Practica No 3 Cinética QuímicaDocumento5 páginasPractica No 3 Cinética QuímicaUriel Cisneros100% (1)
- Normalidad, Molaridad, Molalidad, Soluciones, EjerciciosDocumento6 páginasNormalidad, Molaridad, Molalidad, Soluciones, EjerciciosClaroAún no hay calificaciones
- Análisis de Leche Cruda y Leche PasteurizadaDocumento43 páginasAnálisis de Leche Cruda y Leche PasteurizadaSyg DelgadoAún no hay calificaciones
- LDHDocumento3 páginasLDHLeandro GarnicaAún no hay calificaciones
- DisolutoresDocumento6 páginasDisolutoresMiguelAún no hay calificaciones
- Norma Oficial MexicanaDocumento41 páginasNorma Oficial MexicanaSilvia GarciaAún no hay calificaciones
- El DisolutorDocumento4 páginasEl DisolutorPatty Soncco SonccoAún no hay calificaciones
- Fundamentos y Técnicas de Análisis MicrobiológicosDocumento37 páginasFundamentos y Técnicas de Análisis MicrobiológicosEliana Guillín100% (1)
- DossierDocumento27 páginasDossierpepelalohv100% (2)
- Interferencias Con Las Pruebas de LaboratorioDocumento1 páginaInterferencias Con Las Pruebas de LaboratorioLiz M VillaVqzAún no hay calificaciones
- Especificaciones de Calidad de Los Productos TerminadosDocumento10 páginasEspecificaciones de Calidad de Los Productos TerminadosROSA LUZAún no hay calificaciones
- Análisis Citoquímico de Materia FecalDocumento19 páginasAnálisis Citoquímico de Materia FecalGisela R FrancoAún no hay calificaciones
- Farmacotecnia. Formas Farmaceuticas No EsterilesDocumento21 páginasFarmacotecnia. Formas Farmaceuticas No EsterilesMónica GutierrezAún no hay calificaciones
- Validación de Un Método Por RP-HPLC para La Determinación de Tiocolchicósido en TabletasDocumento7 páginasValidación de Un Método Por RP-HPLC para La Determinación de Tiocolchicósido en TabletasVarinia ZubiletaAún no hay calificaciones
- Transferencia de MétodosDocumento2 páginasTransferencia de MétodosJosahany Castillo50% (2)
- Validacion de Metodos Invima PDFDocumento86 páginasValidacion de Metodos Invima PDFFelix Mendoza100% (1)
- Validacion de Metodos AnaliticosDocumento90 páginasValidacion de Metodos AnaliticosVictoria Gonzales50% (4)
- Tesis MetamizolDocumento93 páginasTesis MetamizolWALDEMAR MARISCAL FLORESAún no hay calificaciones
- InsertoDocumento2 páginasInsertoLenin MorenoAún no hay calificaciones
- 04 Valoracion Enfermera - Patrones Funcionales Salud Gordon 13 MODIFICADOS - DIRAYA-AZAHARDocumento5 páginas04 Valoracion Enfermera - Patrones Funcionales Salud Gordon 13 MODIFICADOS - DIRAYA-AZAHARMaite Alis Carballo AAún no hay calificaciones
- Antecedentes Históricos de La Tecnología FarmacéuticaDocumento5 páginasAntecedentes Históricos de La Tecnología FarmacéuticaAnthony Castro Pacheco100% (2)
- Manual de Procedimientos de Química ClínicaDocumento5 páginasManual de Procedimientos de Química ClínicaMiguel HAún no hay calificaciones
- Anexo 6 Informe 34 OMSDocumento22 páginasAnexo 6 Informe 34 OMSHumberto Colón100% (1)
- Curso Validacion de Metodos AnaliticosDocumento84 páginasCurso Validacion de Metodos Analiticosyocam2100% (1)
- Guía Análisis Toxicológicos 2018Documento91 páginasGuía Análisis Toxicológicos 2018Marino Belisario Baca EspinozaAún no hay calificaciones
- Control de Calidad Interno - PDFDocumento14 páginasControl de Calidad Interno - PDFSergio Giovanni Aguilar PastorAún no hay calificaciones
- Mantenimiento de MedicamentoDocumento36 páginasMantenimiento de MedicamentoCinthia Moreira Vera0% (1)
- Pno Tabletas FinalDocumento10 páginasPno Tabletas FinalCésar Adrián Alavez RamírezAún no hay calificaciones
- CPP o CLC y BPMDocumento8 páginasCPP o CLC y BPMAlluka SacAún no hay calificaciones
- Práctica 1 - Verificación de La BalanzaDocumento5 páginasPráctica 1 - Verificación de La BalanzaJhean Pierre FuentesAún no hay calificaciones
- Tipos de PCRDocumento3 páginasTipos de PCRmagaacastilloAún no hay calificaciones
- Elisa... PresentacionDocumento37 páginasElisa... PresentacionAracelly Gomez MartinezAún no hay calificaciones
- Calificación y Validación PDFDocumento6 páginasCalificación y Validación PDFveronicaAún no hay calificaciones
- Validación de Métodos AnalíticosDocumento68 páginasValidación de Métodos AnalíticosangelaAún no hay calificaciones
- Perfil ReumáticoDocumento22 páginasPerfil ReumáticoElvis Joé Luján RuizAún no hay calificaciones
- Ass Rsa Gu055Documento12 páginasAss Rsa Gu055karen llanosAún no hay calificaciones
- Guía para Bioequivalencia de CofeprisDocumento7 páginasGuía para Bioequivalencia de CofeprisRodolfo Pinto AlmazánAún no hay calificaciones
- MGA-MD1241-procedimientos de Muestreo y Tablas de Inspeccion Por AtributosDocumento23 páginasMGA-MD1241-procedimientos de Muestreo y Tablas de Inspeccion Por AtributosAlejandra LopezAún no hay calificaciones
- Quimica - Volumetria y GravimetriaDocumento4 páginasQuimica - Volumetria y GravimetriaLesly VillegasAún no hay calificaciones
- Abril Nota Tecnica Explicación Validacion LimpiezaDocumento6 páginasAbril Nota Tecnica Explicación Validacion LimpiezacarbouAún no hay calificaciones
- Informe Agente SuspensorDocumento9 páginasInforme Agente SuspensorDaniel Carrillo OrtizAún no hay calificaciones
- XyzDocumento21 páginasXyzJuan Jose Changllio RoasAún no hay calificaciones
- RegulacionesDocumento50 páginasRegulacionesJohan Paredes LeveauAún no hay calificaciones
- Leccion 2 Control de Calidad de Medicamentos PDFDocumento13 páginasLeccion 2 Control de Calidad de Medicamentos PDFEvelyn100% (1)
- BIOEQUIVALENCIA-Y-REGLAMENTO-DE-INTERCAMBIABILIDAD UltimoDocumento10 páginasBIOEQUIVALENCIA-Y-REGLAMENTO-DE-INTERCAMBIABILIDAD UltimoCieloAún no hay calificaciones
- Nom 177 Bioeq FinalDocumento41 páginasNom 177 Bioeq FinalDarla SanchezAún no hay calificaciones
- Act 1-FeumDocumento3 páginasAct 1-FeumLeslie LpbAún no hay calificaciones
- Clenbuterol y AntibioticosDocumento13 páginasClenbuterol y AntibioticosLili Alonso RoqueAún no hay calificaciones
- Bioquimica Clinica 2a Ed - Allan GawDocumento145 páginasBioquimica Clinica 2a Ed - Allan GawOsvaldo Orozco90% (10)
- Articulo Sistemas Terapeuticos Transdermicos (STT)Documento4 páginasArticulo Sistemas Terapeuticos Transdermicos (STT)Lili Alonso RoqueAún no hay calificaciones
- Inmunohistoquimica e InmunoflorescenciaDocumento2 páginasInmunohistoquimica e InmunoflorescenciaLili Alonso RoqueAún no hay calificaciones
- Genetica en Medicina Thompson 7Documento578 páginasGenetica en Medicina Thompson 7Lili Alonso Roque100% (2)
- Bioquimica Clinica 2a Ed - Allan GawDocumento145 páginasBioquimica Clinica 2a Ed - Allan GawOsvaldo Orozco90% (10)
- Preparacion de SolucionesDocumento4 páginasPreparacion de SolucionesLili Alonso RoqueAún no hay calificaciones
- PARA EMPEZAR Guia 1Documento8 páginasPARA EMPEZAR Guia 1Lili Alonso RoqueAún no hay calificaciones
- Capítulo 1 Introducción A La Toxicología 2013-I PDFDocumento11 páginasCapítulo 1 Introducción A La Toxicología 2013-I PDFLili Alonso RoqueAún no hay calificaciones
- Capítulo 1 Introducción A La Toxicología 2013-I PDFDocumento11 páginasCapítulo 1 Introducción A La Toxicología 2013-I PDFLili Alonso RoqueAún no hay calificaciones
- LiguDocumento11 páginasLiguLili Alonso RoqueAún no hay calificaciones
- Capítulo ..3Documento17 páginasCapítulo ..3Julio MartinezAún no hay calificaciones
- Muestreo SistematicoDocumento2 páginasMuestreo SistematicoLili Alonso RoqueAún no hay calificaciones
- El Sistema de Clasificación ATC de Sustancias Farmacéuticas para Uso HumanoDocumento2 páginasEl Sistema de Clasificación ATC de Sustancias Farmacéuticas para Uso HumanoLili Alonso RoqueAún no hay calificaciones
- BGIDocumento11 páginasBGILili Alonso RoqueAún no hay calificaciones
- Quimica de La CoordinaciónDocumento17 páginasQuimica de La CoordinaciónKyra_clik50% (2)
- Efecto Del Vino y Queso en La Sinapsis AdrenérgicaDocumento2 páginasEfecto Del Vino y Queso en La Sinapsis AdrenérgicaLili Alonso Roque100% (1)
- BARBÍTURICOSDocumento1 páginaBARBÍTURICOSJe MolinariAún no hay calificaciones
- Las Presiones Parciales de La Mexcla Del BencenoDocumento2 páginasLas Presiones Parciales de La Mexcla Del BencenoLili Alonso RoqueAún no hay calificaciones
- Tarea4-Fundamentos de Sistemas de ControlDocumento7 páginasTarea4-Fundamentos de Sistemas de ControlLaura Marcela Grajales LópezAún no hay calificaciones
- Concreto Vaciado en SitioDocumento13 páginasConcreto Vaciado en SitioHeriberto Yau BAún no hay calificaciones
- HidrologiaDocumento54 páginasHidrologiaHenry Caleb Nuñez RicanquiAún no hay calificaciones
- Posible Efectos de Los Huracanes Al Turismo en República Dominicana - Practica Trabajo de Final de GradoDocumento26 páginasPosible Efectos de Los Huracanes Al Turismo en República Dominicana - Practica Trabajo de Final de GradoLuis Love CastroAún no hay calificaciones
- Manual DDZ1513 Ultima VersionDocumento106 páginasManual DDZ1513 Ultima VersionDennis Guanilo25% (4)
- H° 1 PDFDocumento82 páginasH° 1 PDFGermán Nicolás Luna MoretaAún no hay calificaciones
- Informe de CalibradorDocumento6 páginasInforme de CalibradorSara AguilarAún no hay calificaciones
- Casquete EsféricoDocumento8 páginasCasquete EsféricopolAún no hay calificaciones
- TEMA: Cantidades Escalares y VectorialesDocumento8 páginasTEMA: Cantidades Escalares y Vectorialesoscaralmario75% (4)
- Universidad Nacional de ChimborazoDocumento7 páginasUniversidad Nacional de ChimborazoMayte MielesAún no hay calificaciones
- Hiperestaticidad Interna y ExternaDocumento9 páginasHiperestaticidad Interna y ExternaCarlos Cercado RubioAún no hay calificaciones
- Preguntas Tipo Test - Fluidos IIDocumento2 páginasPreguntas Tipo Test - Fluidos IIKevyn Đávila BurgaAún no hay calificaciones
- Guia h2s (Coepamm)Documento37 páginasGuia h2s (Coepamm)erafael30Aún no hay calificaciones
- Modelo GrecolatinoDocumento11 páginasModelo GrecolatinohenryAún no hay calificaciones
- ResolucionPr02 2010Documento9 páginasResolucionPr02 2010Carlos BarrazaAún no hay calificaciones
- Caída LibreDocumento8 páginasCaída LibreJosenny Diaz OlivoAún no hay calificaciones
- Informe 2 InorganicaDocumento12 páginasInforme 2 InorganicaLuis FerAún no hay calificaciones
- Anticongelante Shell 2Documento2 páginasAnticongelante Shell 2Karen LizAún no hay calificaciones
- Informe Sensores de TemperaturaDocumento6 páginasInforme Sensores de Temperaturaartherisk0% (1)
- Ensayo La Quimica Es Importante en La Ing IndustrialDocumento1 páginaEnsayo La Quimica Es Importante en La Ing IndustrialKarol Balaguera100% (2)
- Datos CuriososDocumento5 páginasDatos Curiososnatalia50% (2)
- Diagrama de Hierro y CarbonoDocumento8 páginasDiagrama de Hierro y CarbonoRodrigo Gutierrez ChavezAún no hay calificaciones
- Analisis de Aceite ABBDocumento9 páginasAnalisis de Aceite ABBjorgeAún no hay calificaciones
- Dictamen Luis Donaldo ColosioDocumento13 páginasDictamen Luis Donaldo Colosiojulio67% (3)
- CIENCIA Y AMBIENTE Catalogo-Modulos-1y2Documento11 páginasCIENCIA Y AMBIENTE Catalogo-Modulos-1y2Mike HuamanAún no hay calificaciones
- Capítulo 34 CT1 8-06 PDFDocumento3 páginasCapítulo 34 CT1 8-06 PDFRodrigo FigariAún no hay calificaciones
- Capitulo 7 AshraeDocumento37 páginasCapitulo 7 AshraelmelendeziAún no hay calificaciones
- Mapa Conceptual FODocumento1 páginaMapa Conceptual FOJOHAN HABID OROZCO ARAUJO67% (3)
- GUIA Sumatorias y ProgresionesDocumento6 páginasGUIA Sumatorias y ProgresionesbaadthAún no hay calificaciones