También podría gustarte
- Casos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSDe EverandCasos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSCalificación: 4.5 de 5 estrellas4.5/5 (7)
- Cuestionario Grupo 5. Plantas Térmicas 2020-IIDocumento56 páginasCuestionario Grupo 5. Plantas Térmicas 2020-IICesar LimasAún no hay calificaciones
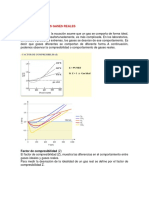
- Energía interna, entalpía y ecuaciones de estado para gases idealesDocumento14 páginasEnergía interna, entalpía y ecuaciones de estado para gases idealesAndres AndradeAún no hay calificaciones
- Marco Teórico GASESDocumento3 páginasMarco Teórico GASESMayraAlejandraVargas100% (2)
- Plantas de conversión térmica 1er examen parcialDocumento19 páginasPlantas de conversión térmica 1er examen parcialLuis F. Estrada GutierrezAún no hay calificaciones
- Gases Reales 20-IiDocumento5 páginasGases Reales 20-IiDanny Palma AyalaAún no hay calificaciones
- Term Odin Á MicaDocumento18 páginasTerm Odin Á MicaJose Pineda RamirezAún no hay calificaciones
- Gases Ideales - David M.himmelblauDocumento15 páginasGases Ideales - David M.himmelblauAriel CristóbalAún no hay calificaciones
- Cuestionario-Segundo ParcialDocumento43 páginasCuestionario-Segundo ParcialJuan Silva LopezAún no hay calificaciones
- PRIMER PARCIAL PLANTAS DE CONVERSIÓN TÉRMICADocumento47 páginasPRIMER PARCIAL PLANTAS DE CONVERSIÓN TÉRMICAJuanPazAún no hay calificaciones
- Cuestionario de PlantasDocumento107 páginasCuestionario de PlantasDaniel Ruge100% (1)
- Sesión 4 Gases Ideales y RealesDocumento8 páginasSesión 4 Gases Ideales y RealesPaola Stefania Villarreal VeraAún no hay calificaciones
- Termodinamica II Tarea 1Documento6 páginasTermodinamica II Tarea 1Alvaro Wara SuárezAún no hay calificaciones
- V. Teoria Cinetica de Los GasesDocumento28 páginasV. Teoria Cinetica de Los Gasesanon_580798559Aún no hay calificaciones
- Ecuaciones gases realesDocumento13 páginasEcuaciones gases realesCristian Ccaso MamaniAún no hay calificaciones
- 2.4 Comportamiento de Gases Reales DJVMDocumento11 páginas2.4 Comportamiento de Gases Reales DJVMalondraAún no hay calificaciones
- Practica #7-2Documento19 páginasPractica #7-2Cynntia Barbara MendozaAún no hay calificaciones
- ActividadesDocumento10 páginasActividadesAndrey LopezAún no hay calificaciones
- Fisicoquimica P2Documento13 páginasFisicoquimica P2Paul Silva GalvezAún no hay calificaciones
- MAT02Documento25 páginasMAT02Edward Tace100% (1)
- Teoría cinética de los gases idealesDocumento17 páginasTeoría cinética de los gases idealesmiguel angel Huaman LezmaAún no hay calificaciones
- Deberes Quimica IndustrialDocumento6 páginasDeberes Quimica IndustrialJoe HualpaAún no hay calificaciones
- Laboratorio 3 de Termodinamica OficialDocumento15 páginasLaboratorio 3 de Termodinamica OficialLuisUsseglioAún no hay calificaciones
- Quimica General IDocumento18 páginasQuimica General ISofía Alvarez HernandezAún no hay calificaciones
- Segundo Trabajo de TermodinamicaDocumento18 páginasSegundo Trabajo de TermodinamicaAlfonzo Antonio Natera OrtegaAún no hay calificaciones
- Cuestionario de PlantasDocumento108 páginasCuestionario de PlantasJuan Silva LopezAún no hay calificaciones
- Laboratorio Integral 4Documento13 páginasLaboratorio Integral 4Analy NolascoAún no hay calificaciones
- Cuestionarios Practica 5Documento8 páginasCuestionarios Practica 5jopisteck01Aún no hay calificaciones
- Cuestionario examen parcial plantas de conversión térmicaDocumento43 páginasCuestionario examen parcial plantas de conversión térmicaAngie GarciaAún no hay calificaciones
- GasesDocumento6 páginasGasesJose Chavez TobarAún no hay calificaciones
- Apuntes Sobre GasesDocumento34 páginasApuntes Sobre GasesAlvaro Ivan Irala Barrios100% (1)
- La Temperatura PDFDocumento8 páginasLa Temperatura PDFFernandoAvilésAún no hay calificaciones
- Clase10 Teoriagases2013aDocumento15 páginasClase10 Teoriagases2013aMauricio RamirezAún no hay calificaciones
- Termodinamica Unidad 1Documento12 páginasTermodinamica Unidad 1Ramm MartinezAún no hay calificaciones
- Ley de los gases ideales y propiedades de mezclas gaseosasDocumento17 páginasLey de los gases ideales y propiedades de mezclas gaseosasAndrimarCorderoAún no hay calificaciones
- Gases Ideales y RealesDocumento13 páginasGases Ideales y RealesLee HansonAún no hay calificaciones
- Antologia Unidad 5Documento29 páginasAntologia Unidad 5Erika Perez Fernando100% (1)
- Unidad IVDocumento20 páginasUnidad IVYubiri Marcano SuarezAún no hay calificaciones
- lab Cp.CvDocumento8 páginaslab Cp.CvvladimirsancheztovarAún no hay calificaciones
- Informe Fiqui 2 1Documento17 páginasInforme Fiqui 2 1BryanAún no hay calificaciones
- ExposiciónDocumento5 páginasExposiciónLuis Jose Aguilar MejiaAún no hay calificaciones
- Sesion 6 - Gases - Resuelto ClaseDocumento32 páginasSesion 6 - Gases - Resuelto ClaseLuis SantiagoAún no hay calificaciones
- Laboratorio n.10Documento7 páginasLaboratorio n.10Matias BarrientosAún no hay calificaciones
- Ecuación de estado gas idealDocumento3 páginasEcuación de estado gas idealLisandro FloresAún no hay calificaciones
- Practica 3 (Actualizada)Documento5 páginasPractica 3 (Actualizada)basurac811Aún no hay calificaciones
- GasesDocumento13 páginasGasesDianaCalderónOyola100% (1)
- Semana 2 FQ Gases RealesDocumento24 páginasSemana 2 FQ Gases RealesJacqueline Machón CamposAún no hay calificaciones
- Estados de agregación y leyes de los gases idealesDocumento9 páginasEstados de agregación y leyes de los gases idealesNorella RiveraAún no hay calificaciones
- Gas RealDocumento23 páginasGas RealAlejandra Adrian TejadaAún no hay calificaciones
- Problemario Termo IPN Septiembre 2015Documento35 páginasProblemario Termo IPN Septiembre 2015Carol Sán0% (1)
- Repasos y Demostraciones en TermodinámicaDocumento16 páginasRepasos y Demostraciones en TermodinámicaJunior PachecoAún no hay calificaciones
- Leyes de los gases ideales CNTPDocumento6 páginasLeyes de los gases ideales CNTPClaudia Mabel FloresAún no hay calificaciones
- Ecuación de Estado de Un Gas IdealDocumento3 páginasEcuación de Estado de Un Gas IdealJipson Uresto100% (1)
- Capítulo 4Documento35 páginasCapítulo 4rofdiazAún no hay calificaciones
- Estados CorrespondientesDocumento16 páginasEstados CorrespondientesCarlos AlarconAún no hay calificaciones
- Unidad III 3.1 3.2 Mecanica de Fluidos y TermodinamicaDocumento5 páginasUnidad III 3.1 3.2 Mecanica de Fluidos y Termodinamicaoliverio cruzAún no hay calificaciones
- Seminarios Primera Parte-2015 PDFDocumento44 páginasSeminarios Primera Parte-2015 PDFVane CarranzaAún no hay calificaciones
- Plantas térmicas - Cuestionario de primer parcialDocumento13 páginasPlantas térmicas - Cuestionario de primer parcialAlejandro Annicherico PuelloAún no hay calificaciones
- Calores Específicos GasesDocumento3 páginasCalores Específicos GasesIradier Amed González CAún no hay calificaciones
- Ciclos de Plantas de Fuerza y Refrigeración Parte IIDocumento27 páginasCiclos de Plantas de Fuerza y Refrigeración Parte IIMaria caballeroAún no hay calificaciones
- Examen 3 Corte QuimicaDocumento3 páginasExamen 3 Corte QuimicaMaria caballeroAún no hay calificaciones
- Transformación de CoordenadasDocumento1 páginaTransformación de CoordenadasMaria caballeroAún no hay calificaciones
- Tarea Fisica Otro 10Documento4 páginasTarea Fisica Otro 10Maria caballeroAún no hay calificaciones
- Trabajo Final de QuimicaDocumento29 páginasTrabajo Final de QuimicaMaria caballeroAún no hay calificaciones
- Orgia Medidas AntiguasDocumento11 páginasOrgia Medidas AntiguasMaria caballeroAún no hay calificaciones
- Ciclos de Plantas de Fuerza y Refrigeración Parte IDocumento17 páginasCiclos de Plantas de Fuerza y Refrigeración Parte IMaria caballeroAún no hay calificaciones
- 2021 - 02 FG Semana - 9Documento85 páginas2021 - 02 FG Semana - 9Darkwolf 2314Aún no hay calificaciones
- Carta PsicrometricaDocumento10 páginasCarta PsicrometricaEduardo Barrantes RamosAún no hay calificaciones
- Aire 1Documento6 páginasAire 1carlos manuel ramirez gonzalezAún no hay calificaciones
- Cuadernillo Matematica Ecr 6to de Primaria 2019Documento12 páginasCuadernillo Matematica Ecr 6to de Primaria 2019jcarlos mejia moquillazauan100% (1)
- Previo Practica 8 Cambio de Entalpía de Fusión Del HieloDocumento2 páginasPrevio Practica 8 Cambio de Entalpía de Fusión Del HieloPedrito Sola100% (1)
- Conversión Temperaturas y EjerciciosDocumento5 páginasConversión Temperaturas y EjerciciosAdriana ReynosoAún no hay calificaciones
- Informe, Biofísica Calor ESP.Documento4 páginasInforme, Biofísica Calor ESP.Meriele AndreaAún no hay calificaciones
- Calculos Propano 2 EtapasDocumento9 páginasCalculos Propano 2 EtapasMaria Victoria Morales GalindezAún no hay calificaciones
- Determinar el calor latente de fusión del hielo UNAM Facultad de QuímicaDocumento4 páginasDeterminar el calor latente de fusión del hielo UNAM Facultad de QuímicaSergio BautistaAún no hay calificaciones
- EntalpíaDocumento21 páginasEntalpíarubenAún no hay calificaciones
- Cuadernillo #3Documento7 páginasCuadernillo #3MILAGRO DE JESUS OSORIO FRIASAún no hay calificaciones
- Noviembre 21 - Bombas de CalorDocumento12 páginasNoviembre 21 - Bombas de CalorJuan Pablo FernandezAún no hay calificaciones
- Transferencia de Calor 2do ParcialDocumento7 páginasTransferencia de Calor 2do ParcialAdriana Estefania Tomalá GómezAún no hay calificaciones
- Anova y LSDDocumento8 páginasAnova y LSDCatalina VargasAún no hay calificaciones
- Calor Especifico de Los MetalesDocumento14 páginasCalor Especifico de Los MetalesHibarixHotaroAún no hay calificaciones
- Vapor de agua enfriado a presión constanteDocumento2 páginasVapor de agua enfriado a presión constanteSergio Rozo Perdomo100% (1)
- Lab 1 Cilo de RefrigeracionDocumento15 páginasLab 1 Cilo de RefrigeracionRolmer García AltamiranoAún no hay calificaciones
- Estados de Liquido Saturado y de Vapor SaturadoDocumento3 páginasEstados de Liquido Saturado y de Vapor SaturadoING. Miguel Angel Sanchez MonzonAún no hay calificaciones
- Equilibrio Material Complemento Iv PDFDocumento10 páginasEquilibrio Material Complemento Iv PDFFernando OrantesAún no hay calificaciones
- Propiedades del aguaDocumento10 páginasPropiedades del aguaMayra AlavezAún no hay calificaciones
- Entropia Energia Libre y EquilibrioDocumento5 páginasEntropia Energia Libre y EquilibrioDiego Alexander Quiliche SánchezAún no hay calificaciones
- Ecuaciones de estado, balances energéticos y de exergía para sistemas termodinámicosDocumento9 páginasEcuaciones de estado, balances energéticos y de exergía para sistemas termodinámicosTotoo SuescunAún no hay calificaciones
- Quinta Parte - Fisica Ii PDFDocumento56 páginasQuinta Parte - Fisica Ii PDFSergio FernandezAún no hay calificaciones
- Primer Parcial (Enunciados) PDFDocumento4 páginasPrimer Parcial (Enunciados) PDFAilec GuarayoAún no hay calificaciones
- K Del IBRD 1280Documento2 páginasK Del IBRD 1280angelAún no hay calificaciones
- PsicrometriaDocumento33 páginasPsicrometriaRoy Neyell YtoAún no hay calificaciones
- Primer Principio de La TermodinámicaDocumento23 páginasPrimer Principio de La TermodinámicaMaria Fernanda Mendez SalcedAún no hay calificaciones
- 0873716001588099124 (2)Documento2 páginas0873716001588099124 (2)andres delgadoAún no hay calificaciones
- Practica 1 Ciclo de Refrigeración Por Compresión de VaporDocumento8 páginasPractica 1 Ciclo de Refrigeración Por Compresión de VaporMilton BermudezAún no hay calificaciones
- Resolución 2DO PARCIAL 2 de Julio F53 Horario 657 Tema ADocumento5 páginasResolución 2DO PARCIAL 2 de Julio F53 Horario 657 Tema Acoco graz0% (1)