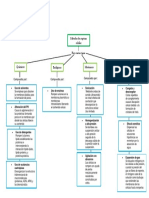
También podría gustarte
- Practica HELMINTOLOGIADocumento5 páginasPractica HELMINTOLOGIAkaren lhzAún no hay calificaciones
- Practica Acido AceticoDocumento26 páginasPractica Acido AceticoIrma Peralta CastroAún no hay calificaciones
- Tren de Fuerza CargadorDocumento5 páginasTren de Fuerza CargadorVc Franki0% (1)
- Aplicaciones de Las Ecs. de Variables Separables.Documento3 páginasAplicaciones de Las Ecs. de Variables Separables.Osvaldo Reyes MagallanesAún no hay calificaciones
- Capitulo 4 - Equilibrio QuimicoDocumento37 páginasCapitulo 4 - Equilibrio QuimicoCRISTHIAN ANDREY PINZON ESTEVEZAún no hay calificaciones
- Cuestionario Enzimas InmovilizadasDocumento5 páginasCuestionario Enzimas InmovilizadasPatricia AlvarengaAún no hay calificaciones
- Area 9 Ritmos de CrecimientoDocumento8 páginasArea 9 Ritmos de CrecimientoKenia CondeAún no hay calificaciones
- La Energía Libre de GibbsDocumento16 páginasLa Energía Libre de GibbsYairy Bermont M BAún no hay calificaciones
- Contacto Directo de Dos Fases InmisciblesDocumento4 páginasContacto Directo de Dos Fases InmisciblesVanesa Chugñay33% (3)
- BioprocesosDocumento8 páginasBioprocesosYadira Gutiérrez ZavalaAún no hay calificaciones
- Caracteristicas KPDocumento3 páginasCaracteristicas KPjcccp941016Aún no hay calificaciones
- Reporte Final Laboratorio Integral 2Documento32 páginasReporte Final Laboratorio Integral 2Raul AlegriaAún no hay calificaciones
- Analisis Modelos Crecimiento MoDocumento21 páginasAnalisis Modelos Crecimiento MoMayte Guerrero100% (1)
- Tema 1 - Fisicoquimica IDocumento117 páginasTema 1 - Fisicoquimica IChristian GerardoAún no hay calificaciones
- Conservacion de Energia Calorifica y Calor EspecificoDocumento7 páginasConservacion de Energia Calorifica y Calor EspecificoEdy Ticahuanca AnccoAún no hay calificaciones
- Hoja-De-Seguridad Canela MolidaDocumento2 páginasHoja-De-Seguridad Canela MolidaCesar MoralesAún no hay calificaciones
- Cuestionario No2 Isoterma de Adsorción. 2021Documento4 páginasCuestionario No2 Isoterma de Adsorción. 2021Ingrid Yaneth Chag SimónAún no hay calificaciones
- Tunel de SecadoDocumento7 páginasTunel de SecadoIsabella GonzalezAún no hay calificaciones
- Cinética y Termodinámica de Los BioprocesosDocumento53 páginasCinética y Termodinámica de Los Bioprocesoseli eliasAún no hay calificaciones
- Vit B2Documento4 páginasVit B2Natalia LeonAún no hay calificaciones
- Enunciados BalanceDocumento4 páginasEnunciados BalanceSalomé MeloAún no hay calificaciones
- Analisis Plastico de BinghamDocumento2 páginasAnalisis Plastico de BinghamIvo SegoviaAún no hay calificaciones
- Glucólisis (Ruta Embden-Meyerhof)Documento26 páginasGlucólisis (Ruta Embden-Meyerhof)José AlfredoAún no hay calificaciones
- CUESTIONARIO QuimicaDocumento6 páginasCUESTIONARIO Quimicagregory pardoAún no hay calificaciones
- Taller Equilibrio - QuimicoDocumento2 páginasTaller Equilibrio - QuimicoMaria Camila Alvarez100% (1)
- Propiedades Coligativas ExposiciónDocumento23 páginasPropiedades Coligativas ExposiciónRichard JimperAún no hay calificaciones
- Taller 2 de Transferencia de CalorDocumento2 páginasTaller 2 de Transferencia de CalorJuanCarlosAún no hay calificaciones
- Bioprocesos CuestionarioDocumento7 páginasBioprocesos CuestionarioDaniel LoorAún no hay calificaciones
- Biosintesis de DesoxirribonucleotidosDocumento23 páginasBiosintesis de DesoxirribonucleotidosingristabAún no hay calificaciones
- Actividad de AguaDocumento10 páginasActividad de AguaJavier Riqueros De FinaAún no hay calificaciones
- Preguntas LaboratorioDocumento7 páginasPreguntas LaboratorioMonica OramasAún no hay calificaciones
- Taller LabDocumento9 páginasTaller LabGiuliana Guerra SalazarAún no hay calificaciones
- Enzimas InmovilizadorasDocumento16 páginasEnzimas InmovilizadorasNiñoGonzalesBenitoAún no hay calificaciones
- Difusion de LiquidosDocumento6 páginasDifusion de LiquidosnznoAún no hay calificaciones
- Principios de Analisis QuimicoDocumento6 páginasPrincipios de Analisis Quimicoandreagonzalez15Aún no hay calificaciones
- Informe de FisicoquimicaDocumento19 páginasInforme de FisicoquimicaLuis Alberto Orbegoso HuancaAún no hay calificaciones
- 6 Lih + 2 Bcl3 B2H6 + 6 LiclDocumento2 páginas6 Lih + 2 Bcl3 B2H6 + 6 LiclWilfredoRivera100% (1)
- Reporte 1Documento12 páginasReporte 1Kevin MirandaAún no hay calificaciones
- Ejercicio Balance de MasaDocumento1 páginaEjercicio Balance de MasaAdrihanaPaladinesAún no hay calificaciones
- Equilibrio QuimicoDocumento8 páginasEquilibrio QuimicoDennis LinoAún no hay calificaciones
- Equilibrio QuímicoDocumento24 páginasEquilibrio QuímicoCarolina PeñúñuriAún no hay calificaciones
- Informe 3 - Calor de NeutralizacionDocumento13 páginasInforme 3 - Calor de NeutralizacionAlex Ostos AlvaAún no hay calificaciones
- Metodos de Ruptura CelularDocumento1 páginaMetodos de Ruptura CelularNay BautistaAún no hay calificaciones
- Procesos de Transferencia en BiorreactoresDocumento6 páginasProcesos de Transferencia en BiorreactoresAxel HernándezAún no hay calificaciones
- Resumen de Adición de Radicales y Reacciones.Documento13 páginasResumen de Adición de Radicales y Reacciones.Javier Aldair Ruiz AmadorAún no hay calificaciones
- Intercambiador Tubos y Coraza EcuaciònesDocumento8 páginasIntercambiador Tubos y Coraza EcuaciònesAndres Felipe GutierrezAún no hay calificaciones
- Efecto de Factores Ambientales en La Curva de CrecimientoDocumento2 páginasEfecto de Factores Ambientales en La Curva de CrecimientoSilvana StarAún no hay calificaciones
- Energia y GlucolisisDocumento12 páginasEnergia y GlucolisishomeroAún no hay calificaciones
- Taller 4Documento6 páginasTaller 4Sebastian CandoAún no hay calificaciones
- Clase SedimentacionDocumento26 páginasClase Sedimentacionvvivizfv0% (1)
- Evaporador de Simple EfectoDocumento5 páginasEvaporador de Simple EfectoJose Kalvin RojasAún no hay calificaciones
- SolubilidadDocumento8 páginasSolubilidadJosman PriteAún no hay calificaciones
- Informe de Espontaneidad y EquilibrioDocumento6 páginasInforme de Espontaneidad y EquilibrioHERNAN ENRIQUE NORIEGA BARROSAún no hay calificaciones
- PRACTICA N 4 BioquimicaDocumento5 páginasPRACTICA N 4 BioquimicaBaciliaMila100% (1)
- Regla 3 ComponentesDocumento2 páginasRegla 3 ComponentesPamela Sanhueza JaraAún no hay calificaciones
- Identificación de ProteínasDocumento7 páginasIdentificación de ProteínasjaliAún no hay calificaciones
- Energía Libre PDFDocumento51 páginasEnergía Libre PDFevizcardo4617Aún no hay calificaciones
- Energa LibreDocumento65 páginasEnerga LibreElías Raphael Rubilar BugueñoAún no hay calificaciones
- Energía LibreDocumento65 páginasEnergía LibreaaasAún no hay calificaciones
- 4.energa Libre ClasesDocumento64 páginas4.energa Libre ClasesJuan Antonio CarlosAún no hay calificaciones
- Clase Cap 1.7 Entropia 2-GIbbsDocumento7 páginasClase Cap 1.7 Entropia 2-GIbbsmiguel EncarnacionAún no hay calificaciones
- 016 Electrostatica OkDocumento2 páginas016 Electrostatica OkEdithYolandaColquehuancaYujraAún no hay calificaciones
- Pets-Lb-Ins-03 Tendido de Tuberia Corrugada PVC y Cable Utp para CamaraDocumento9 páginasPets-Lb-Ins-03 Tendido de Tuberia Corrugada PVC y Cable Utp para Camararoberto capacAún no hay calificaciones
- NTC - 2047 2002 1° Dibujo SimbolosDocumento16 páginasNTC - 2047 2002 1° Dibujo SimbolosMaliton QuimbayoAún no hay calificaciones
- SAIDI Valvulas Mariposa SUFADocumento12 páginasSAIDI Valvulas Mariposa SUFARicardo SGAún no hay calificaciones
- Ejercicio Modulo 2Documento4 páginasEjercicio Modulo 2serviciosgenerales.hen2023Aún no hay calificaciones
- Desarrollo Comunitario y Protección de Los Recursos Humanos1Documento19 páginasDesarrollo Comunitario y Protección de Los Recursos Humanos1Aguin Ial100% (1)
- Memoria e Calculo Modulo AdministracionDocumento101 páginasMemoria e Calculo Modulo AdministracionErik TrujilloAún no hay calificaciones
- Serie 4 - Cursillo Qca 2014 PDocumento19 páginasSerie 4 - Cursillo Qca 2014 PHoracio GorosteguiAún no hay calificaciones
- Simulacion Campo MagneticoDocumento4 páginasSimulacion Campo MagneticolandetaAún no hay calificaciones
- Programa de Financiamiento para Emprendedor A Través de La Banca ComercialDocumento4 páginasPrograma de Financiamiento para Emprendedor A Través de La Banca Comercialfany pmAún no hay calificaciones
- Leica EG1160 Manual ESDocumento32 páginasLeica EG1160 Manual ESandreapasitos8340100% (1)
- Subestaciones I v3Documento69 páginasSubestaciones I v3SALVADORAún no hay calificaciones
- Ejemplo Libro de COMPRAS ABRIL 2019Documento12 páginasEjemplo Libro de COMPRAS ABRIL 2019d verdugocAún no hay calificaciones
- Solucion QuimicaDocumento9 páginasSolucion QuimicaLuisa MaloAún no hay calificaciones
- Instructivo Operacion CalderaDocumento3 páginasInstructivo Operacion CalderaCarlosQuintoAún no hay calificaciones
- Guia - Ley de BoyleDocumento5 páginasGuia - Ley de BoyleMiguelAngelAntezanaVergaraAún no hay calificaciones
- Literal b1 Directorio de La Institucion0387731001602012590Documento104 páginasLiteral b1 Directorio de La Institucion0387731001602012590dfarevaloAún no hay calificaciones
- Reporte MantenimientoDocumento1 páginaReporte MantenimientoEstebanAún no hay calificaciones
- Libro Del Sector CañeroDocumento226 páginasLibro Del Sector CañeroFrancisco Javier Medina SorianoAún no hay calificaciones
- Certificado SPATDocumento8 páginasCertificado SPATalbertoAún no hay calificaciones
- Prueba Ciencias 7° Unidad 2Documento4 páginasPrueba Ciencias 7° Unidad 2Luz AndreaAún no hay calificaciones
- Cargadores de La Serie 9SDocumento64 páginasCargadores de La Serie 9SEmilio Giraldo100% (2)
- Que El BIL y El BSL Se Administren para Las Condiciones Del Nivel Del MarDocumento8 páginasQue El BIL y El BSL Se Administren para Las Condiciones Del Nivel Del MarJesusVilcaYepezAún no hay calificaciones
- Act Apren4 JMSODocumento13 páginasAct Apren4 JMSOMario OlivaresAún no hay calificaciones
- Curso de Electricidad Del Automovil, Simbologia PDFDocumento9 páginasCurso de Electricidad Del Automovil, Simbologia PDFalix1977Aún no hay calificaciones
- Rubber ElasticityDocumento39 páginasRubber ElasticityingdayronmrAún no hay calificaciones
- Compensacion Fuzzy LogicDocumento7 páginasCompensacion Fuzzy LogicPedro LeonelAún no hay calificaciones
- Catalogo HokerDocumento14 páginasCatalogo HokerFabian Antonio RoldanAún no hay calificaciones
- Era PrecambricaDocumento14 páginasEra PrecambricaAristoteles CarreñoAún no hay calificaciones