También podría gustarte
- Control de Calidad Interno de HematologiaDocumento5 páginasControl de Calidad Interno de HematologiaJenny Maria Osorio Sevilla86% (7)
- SinusitisDocumento3 páginasSinusitisRicardo AndrésAún no hay calificaciones
- EpicrisisDocumento2 páginasEpicrisisRicardo AndrésAún no hay calificaciones
- Sindrome de Incontinencia UrinariaDocumento31 páginasSindrome de Incontinencia UrinariaRicardo Andrés100% (1)
- Clasificación de Los ÁngelesDocumento5 páginasClasificación de Los ÁngelesJorge Rosas100% (1)
- Tiroidectomia TotalDocumento3 páginasTiroidectomia TotalRicardo AndrésAún no hay calificaciones
- Preguntas y Respuestas UnachDocumento1 páginaPreguntas y Respuestas UnachRicardo AndrésAún no hay calificaciones
- EpicrisisDocumento6 páginasEpicrisisRicardo AndrésAún no hay calificaciones
- Diseccion Inguinal IzquierdaDocumento3 páginasDiseccion Inguinal IzquierdaRicardo AndrésAún no hay calificaciones
- Parotidectomia SuperficialDocumento3 páginasParotidectomia SuperficialRicardo AndrésAún no hay calificaciones
- Tiroidectomia Total + Diseccion Cervical Funcional IzquierdaDocumento10 páginasTiroidectomia Total + Diseccion Cervical Funcional IzquierdaRicardo AndrésAún no hay calificaciones
- Ecu Habil 01Documento13 páginasEcu Habil 01Ricardo AndrésAún no hay calificaciones
- Síndromes GeriátricosDocumento2 páginasSíndromes GeriátricosRicardo AndrésAún no hay calificaciones
- Algoritmo en La Displasia Del Desarrollo de Las CaderasDocumento52 páginasAlgoritmo en La Displasia Del Desarrollo de Las CaderasRicardo AndrésAún no hay calificaciones
- Clasificacion de FracturasDocumento27 páginasClasificacion de FracturasRicardo AndrésAún no hay calificaciones
- Apa - Pediatria Ii - A - Grupo E2 - Leucemias en Pediatría - Frecuencia y Factores Asociados A Las Leucemias en PediatriaDocumento29 páginasApa - Pediatria Ii - A - Grupo E2 - Leucemias en Pediatría - Frecuencia y Factores Asociados A Las Leucemias en Pediatriabenito cameloAún no hay calificaciones
- Iper Construccion CivilDocumento7 páginasIper Construccion Civilgabriel chavezaguirre50% (2)
- Plan de Acción Jornada Nacional de Vacunación - Abri 2021lDocumento6 páginasPlan de Acción Jornada Nacional de Vacunación - Abri 2021lvacunacion vacunacionAún no hay calificaciones
- Difrencia Entre Capa Simple y Compleja Actv-Nº09Documento2 páginasDifrencia Entre Capa Simple y Compleja Actv-Nº09Gabriela LuceroAún no hay calificaciones
- Trastorno Depresivo MayorDocumento19 páginasTrastorno Depresivo MayorMilagros Mendoza100% (1)
- Banco Preguntas Practica1Documento6 páginasBanco Preguntas Practica1Diana SvlAún no hay calificaciones
- Normativa 121 ExposiciónDocumento6 páginasNormativa 121 ExposiciónElvin AvilésAún no hay calificaciones
- AGNOSIASDocumento9 páginasAGNOSIASJulieta SuarezAún no hay calificaciones
- CORRECCIONES. Augusto Querales #30, Ariadna Santaella 19. 5to Año BDocumento20 páginasCORRECCIONES. Augusto Querales #30, Ariadna Santaella 19. 5to Año BAugusto Arturo Querales AmayaAún no hay calificaciones
- Spa MenuDocumento33 páginasSpa Menujose lopezAún no hay calificaciones
- Parcial Semio DefinitivoDocumento34 páginasParcial Semio DefinitivoJulian Oliveros velasquezAún no hay calificaciones
- CV Eduardo SotoDocumento2 páginasCV Eduardo SotoCésar Calisto VegaAún no hay calificaciones
- Cap 33 AlergiasDocumento8 páginasCap 33 AlergiasAndreaCostalesAún no hay calificaciones
- Resumen PacientesDocumento45 páginasResumen PacientesDaniela Vargas GómezAún no hay calificaciones
- Geriatria Tarea 1Documento4 páginasGeriatria Tarea 1Aylin AlvarezAún no hay calificaciones
- Pitiriasis VersicolorDocumento21 páginasPitiriasis VersicolorChristianParedesAún no hay calificaciones
- Ano y RectoDocumento32 páginasAno y RectoJuan Carlos Marcos EnriquezAún no hay calificaciones
- Curso de Interacciones Medicamentosas-1Documento105 páginasCurso de Interacciones Medicamentosas-1mmondaca1974Aún no hay calificaciones
- Candida Auris, Nuevo Hongo Patogeno MultirresistenteDocumento19 páginasCandida Auris, Nuevo Hongo Patogeno MultirresistenteRICARDO SOTO AGUDELOAún no hay calificaciones
- Cronograma de CapacitacionDocumento3 páginasCronograma de CapacitacionTannia ArcosAún no hay calificaciones
- Caso 10Documento2 páginasCaso 10Alejandro RíosAún no hay calificaciones
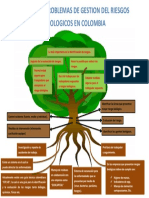
- Arbol de Problemas Riesgos BiologicosDocumento1 páginaArbol de Problemas Riesgos BiologicosVanessa Lopera71% (7)
- CATALOGO ORIFLAME PERU 14 2021 (Vence 22 OCT) WhatsApp 994323931Documento69 páginasCATALOGO ORIFLAME PERU 14 2021 (Vence 22 OCT) WhatsApp 994323931Catálogos MujerAún no hay calificaciones
- Programa Académico XX Congreso Multidisciplinario Issste Victoria 2022 OptDocumento7 páginasPrograma Académico XX Congreso Multidisciplinario Issste Victoria 2022 OptJuanAún no hay calificaciones
- Cuidados de Enfermeria Al Paciente Con Enfermedades CrónicoDocumento38 páginasCuidados de Enfermeria Al Paciente Con Enfermedades CrónicoIrma Muñoz50% (4)
- Protectores de Mucosa, Laxantes, CarticosDocumento16 páginasProtectores de Mucosa, Laxantes, CarticosLaura DíazAún no hay calificaciones
- Daño Mediante ToxinasDocumento53 páginasDaño Mediante ToxinasCamilo Agudelo MuñozAún no hay calificaciones
- Bitacora N°233 - Darien Flores 06-07-2023 DíaDocumento2 páginasBitacora N°233 - Darien Flores 06-07-2023 Díahospital hv heavAún no hay calificaciones