También podría gustarte
- Presentacion DcaDocumento17 páginasPresentacion Dcabrayan felipe de hoyos jimenezAún no hay calificaciones
- 0 Modelo TemperaturaDocumento3 páginas0 Modelo TemperaturaLuis AlccaAún no hay calificaciones
- Convección en CilindrosDocumento38 páginasConvección en CilindrosBrenda Lizbeth Guillen SustaitaAún no hay calificaciones
- Otros Ejercicios Resueltos de Intercambiadores de CalorDocumento31 páginasOtros Ejercicios Resueltos de Intercambiadores de CalorJavier GarridoAún no hay calificaciones
- IV. Diseño de Bloques Completos Al AzarDocumento18 páginasIV. Diseño de Bloques Completos Al Azarjorge QuillaAún no hay calificaciones
- EJERCITARIO7Documento12 páginasEJERCITARIO7ADRB1804Aún no hay calificaciones
- Torsion 1-1Documento26 páginasTorsion 1-1Andres Luis Barreto HidalgoAún no hay calificaciones
- Semana 3Documento3 páginasSemana 3Mirella Adriazola SotoAún no hay calificaciones
- Examen II FS0310 II 2023-3 DefinitivoDocumento9 páginasExamen II FS0310 II 2023-3 DefinitivoSebastian ChacónAún no hay calificaciones
- Laboratorio de Transferencia de Calor 2Documento12 páginasLaboratorio de Transferencia de Calor 2Digna Bettin CuelloAún no hay calificaciones
- Informe Mec Fluidos - 1Documento3 páginasInforme Mec Fluidos - 1Walter PampaloniAún no hay calificaciones
- Sesion 7 Reactor de Flujo en PistonDocumento24 páginasSesion 7 Reactor de Flujo en PistonmarioviusAún no hay calificaciones
- Torsión ClaseDocumento21 páginasTorsión ClaseLuz Yamile PatarroyoAún no hay calificaciones
- Capitulos Iv y VDocumento15 páginasCapitulos Iv y VAmvlarieel SolrosAún no hay calificaciones
- Ejercicios y Problemas de Un MasDocumento5 páginasEjercicios y Problemas de Un MasGranados Bello Jorge FernandoAún no hay calificaciones
- Capítulo Analisis en El TiempoDocumento66 páginasCapítulo Analisis en El TiempoPepe El ToroAún no hay calificaciones
- (Razones Trigonométricas de Ángulos Agudos I PDFDocumento15 páginas(Razones Trigonométricas de Ángulos Agudos I PDFEduardo Pérez SosaAún no hay calificaciones
- Guia Iea Examen ExensDocumento32 páginasGuia Iea Examen ExensAlonso HernandezAún no hay calificaciones
- Trigonometria IiDocumento10 páginasTrigonometria IiAugusto Sánchez CardozoAún no hay calificaciones
- R.T. de Angulos Agudos ANDYDocumento4 páginasR.T. de Angulos Agudos ANDYAndy HuillcaAún no hay calificaciones
- Trigonometria - A7Documento11 páginasTrigonometria - A7Augusto Sánchez CardozoAún no hay calificaciones
- Guia Lab PRQ3209Documento6 páginasGuia Lab PRQ3209Vier YucraAún no hay calificaciones
- Trigo - Identidades TrigoDocumento7 páginasTrigo - Identidades TrigoJose Ernesto Maza RuizAún no hay calificaciones
- P 2Documento2 páginasP 2migan1993Aún no hay calificaciones
- Derrame de Petróleo en ExpansiónDocumento3 páginasDerrame de Petróleo en Expansiónwilmar100% (1)
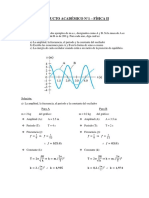
- PRODUCTO ACADÉMICO N°1 - FÍsica IIDocumento10 páginasPRODUCTO ACADÉMICO N°1 - FÍsica IIJean Paul Castro BuendiaAún no hay calificaciones
- ABC Reactores 2aed C10Documento45 páginasABC Reactores 2aed C10OskarAún no hay calificaciones
- Sistema HíbridoDocumento7 páginasSistema HíbridoJesus Javier Albarran SantiagoAún no hay calificaciones
- Razones Trigonometricas de Un AnguloDocumento4 páginasRazones Trigonometricas de Un AnguloJimy LopezAún no hay calificaciones
- Texto Trigonometria PDFDocumento89 páginasTexto Trigonometria PDFPabloHuancaMendezAún no hay calificaciones
- Separata DBCA y Pruebas de Medias MEIDocumento10 páginasSeparata DBCA y Pruebas de Medias MEIJuan Carlos GorizhiñoAún no hay calificaciones
- Labo 4 Filtros DigitalesDocumento13 páginasLabo 4 Filtros DigitaleswilliamAún no hay calificaciones
- S14 Raz Trig Ang AgudosDocumento6 páginasS14 Raz Trig Ang AgudosSaul JaboAún no hay calificaciones
- NocionesFiltradoAnalogicoASS UTNDocumento20 páginasNocionesFiltradoAnalogicoASS UTNAlga SebasAún no hay calificaciones
- Razones TrigonométricasDocumento4 páginasRazones TrigonométricasLuis Adalberto Zapata Valdiviezo100% (2)
- Teoria de RebobinadosDocumento26 páginasTeoria de RebobinadosMiguel Angel HerbasAún no hay calificaciones
- Aleta AnularDocumento2 páginasAleta AnularKike CarreteroAún no hay calificaciones
- Ficha N°7-Razones TrigonometricasDocumento9 páginasFicha N°7-Razones TrigonometricasRafael Bayona OsorioAún no hay calificaciones
- Ejercicios Oscilaciones ClaseDocumento2 páginasEjercicios Oscilaciones ClaseJoséAún no hay calificaciones
- T SUNI Dir Sem11Documento2 páginasT SUNI Dir Sem11Andy Cartolin RosalesAún no hay calificaciones
- Diseño Cuadrado Latino y Grecolatino.Documento13 páginasDiseño Cuadrado Latino y Grecolatino.calebAún no hay calificaciones
- Villa Morales Paola LAB1 PRQ3209Documento18 páginasVilla Morales Paola LAB1 PRQ3209Paola Villa MoralesAún no hay calificaciones
- Castellan 4Documento14 páginasCastellan 4Margarita LlantenAún no hay calificaciones
- Modelo Cuadrado Latino y Ejercicio 4Documento5 páginasModelo Cuadrado Latino y Ejercicio 4Maria TorrealbaAún no hay calificaciones
- Práctica 6 Determinación Experimental de La Frecuencia Natural de Un Péndulo de TorsiónDocumento9 páginasPráctica 6 Determinación Experimental de La Frecuencia Natural de Un Péndulo de TorsiónAlainIslasAún no hay calificaciones
- Razones Trigonométricas de Ángulos AgudosDocumento9 páginasRazones Trigonométricas de Ángulos AgudosJimy LopezAún no hay calificaciones
- SET PREGUNTAS PRUEBA 26 Mayo 2021Documento28 páginasSET PREGUNTAS PRUEBA 26 Mayo 2021alyaAún no hay calificaciones
- Problemas ResueltosDocumento38 páginasProblemas ResueltosVictor Aldunate100% (1)
- 6) Flujo NO IDEAL - m22Documento20 páginas6) Flujo NO IDEAL - m22Guillermo FerrerAún no hay calificaciones
- Formulario de ConducciónDocumento7 páginasFormulario de ConducciónAlexander AlulemaAún no hay calificaciones
- Material 02 PDFDocumento9 páginasMaterial 02 PDFMey Meier HumallaAún no hay calificaciones
- Material 02 PDFDocumento9 páginasMaterial 02 PDFMey Meier HumallaAún no hay calificaciones
- Compensadores y Bode PDFDocumento40 páginasCompensadores y Bode PDFSONIA ANABEL JAQUEZ OLVERAAún no hay calificaciones
- 3 Procesos de 1er OrdenDocumento20 páginas3 Procesos de 1er OrdenFelipe MonteroAún no hay calificaciones
- Sistemas Dinámicos de Primer OrdenDocumento7 páginasSistemas Dinámicos de Primer OrdenIVAN ALFREDO COTA NINAAún no hay calificaciones
- POTABLE EXAMEN 01 PRACTICO 01-2020 Floculación SOLUCIONDocumento4 páginasPOTABLE EXAMEN 01 PRACTICO 01-2020 Floculación SOLUCIONluis eduardo guerrero sotoAún no hay calificaciones
- Apuntes de TrigonometriaDocumento13 páginasApuntes de TrigonometriaJumovagmail.com Jumova01Aún no hay calificaciones
- Formulas Diseño en Cudrado LatinoDocumento7 páginasFormulas Diseño en Cudrado LatinoDonald ContrerasAún no hay calificaciones
- Semana 5 - 2015exDocumento10 páginasSemana 5 - 2015exAdolfo TBAún no hay calificaciones
- Canta Lengua Jubilosa AcordesDocumento2 páginasCanta Lengua Jubilosa AcordesCharles Albert Esquivel Espinoza100% (1)
- Informe 4 MoliendaDocumento4 páginasInforme 4 MoliendaDiego RoqueAún no hay calificaciones
- Caso Clinico AlturasDocumento2 páginasCaso Clinico AlturasMaria De Jesus Blanco HinestrozaAún no hay calificaciones
- Tema 2Documento2 páginasTema 2Laura Roman DelgadoAún no hay calificaciones
- Practica 1 RadiadoresDocumento7 páginasPractica 1 RadiadoresYamile Yael HilarioAún no hay calificaciones
- CAMPAÑA OPTICAL CENTER - CompressedDocumento21 páginasCAMPAÑA OPTICAL CENTER - CompressedEmanuel HrdzAún no hay calificaciones
- Instrumentos FinancierosDocumento49 páginasInstrumentos FinancierosVale OchoaAún no hay calificaciones
- 2.estudio Mecanica de SuelosDocumento23 páginas2.estudio Mecanica de SuelosAlinser Alcántara CabrejosAún no hay calificaciones
- Proyecto Productivo Comunidad PoromanaDocumento20 páginasProyecto Productivo Comunidad PoromanaAndy Arón Alexander Infante OjedaAún no hay calificaciones
- Clinica MuerteDocumento2 páginasClinica MuerteLenin Hernandez RuizAún no hay calificaciones
- Perú-Jennifer LudeñaDocumento15 páginasPerú-Jennifer LudeñaJorge De La CruzAún no hay calificaciones
- Ficha Tecnica AminoacidosDocumento14 páginasFicha Tecnica AminoacidosalbertoAún no hay calificaciones
- Tema 14 Ruta Ciclo de Krebs. Autor Alexander Sanchez Lamas.Documento19 páginasTema 14 Ruta Ciclo de Krebs. Autor Alexander Sanchez Lamas.Alexander SanchezAún no hay calificaciones
- TAREA 1 - Yesenia OcupaDocumento12 páginasTAREA 1 - Yesenia Ocupaedward andres adaime muozAún no hay calificaciones
- Devocionario Franciscano PDFDocumento118 páginasDevocionario Franciscano PDFsimingtonAún no hay calificaciones
- PPT2 - Marketing DigitalDocumento35 páginasPPT2 - Marketing DigitalAngie DerteanoAún no hay calificaciones
- Goldschmidt y Alexy. Corsi e Ricorsi Del Integrativismo Jurídico. cf140845f1Documento10 páginasGoldschmidt y Alexy. Corsi e Ricorsi Del Integrativismo Jurídico. cf140845f1Leandro AcostaAún no hay calificaciones
- EmprendimientoDocumento16 páginasEmprendimientoAdriel CandiaAún no hay calificaciones
- Analisis Del Lugar ADocumento45 páginasAnalisis Del Lugar ADego F. Palomino HerreraAún no hay calificaciones
- GM La Teoría de Los ColibríesDocumento18 páginasGM La Teoría de Los ColibríesJohn Fredy Medina33% (3)
- PosilleroDocumento22 páginasPosilleroLeonardo Stiven Borja ParedesAún no hay calificaciones
- Solución Guia 3 - Modulo - 1 - Unidad - 2 - Cesar - CamachoDocumento25 páginasSolución Guia 3 - Modulo - 1 - Unidad - 2 - Cesar - CamachoEstefani CamachoAún no hay calificaciones
- Psicología de La Educación Historia de La Psicología Del La EducaciónDocumento2 páginasPsicología de La Educación Historia de La Psicología Del La EducaciónDaiana Pérez GiménezAún no hay calificaciones
- Presentación CLASE 2 BASESDocumento21 páginasPresentación CLASE 2 BASESCarmen PadillaAún no hay calificaciones
- Recetas Con Avena y HuevoDocumento4 páginasRecetas Con Avena y HuevoFacundo TremsalAún no hay calificaciones
- Programa Indicativo BASES FILOSÓFICAS Agosto 11Documento23 páginasPrograma Indicativo BASES FILOSÓFICAS Agosto 11rositariverarueda440% (1)
- Romeo y Julieta-Guion - para CombinarDocumento7 páginasRomeo y Julieta-Guion - para CombinarGénesisAún no hay calificaciones
- Reinera ProfesionalDocumento15 páginasReinera Profesionalneldon mendozaAún no hay calificaciones
- Cambios Que Traen SanidadDocumento178 páginasCambios Que Traen SanidadConsuelo Samuel100% (1)
- Evidencia 1Documento7 páginasEvidencia 1alex rocha100% (1)