También podría gustarte
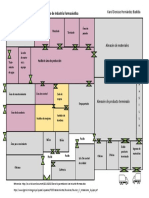
- Plano-Industria Farmacéutica - Hernández Bastida KarolDocumento1 páginaPlano-Industria Farmacéutica - Hernández Bastida KarolKarol Denisse Hernández Bastida86% (7)
- SERIE DE PROBLEMAS, LABORATORIO INTEGRATIVO I CE-II-Hernández Bastida KarolDocumento13 páginasSERIE DE PROBLEMAS, LABORATORIO INTEGRATIVO I CE-II-Hernández Bastida KarolKarol Denisse Hernández BastidaAún no hay calificaciones
- Fitovigilancia 26072020Documento78 páginasFitovigilancia 26072020Karol Denisse Hernández BastidaAún no hay calificaciones
- Serie B - 2021-HernándezBastidaKarolDocumento8 páginasSerie B - 2021-HernándezBastidaKarolKarol Denisse Hernández BastidaAún no hay calificaciones
- Sistema CAPADocumento1 páginaSistema CAPAKarol Denisse Hernández BastidaAún no hay calificaciones
- Diagrama de Flujo-Obtención de PlasmaDocumento1 páginaDiagrama de Flujo-Obtención de PlasmaKarol Denisse Hernández BastidaAún no hay calificaciones
- Preguntas FarmacologiaDocumento6 páginasPreguntas FarmacologiaAaron GarciiaAún no hay calificaciones
- Presentación 1Documento2 páginasPresentación 1irene_amnellAún no hay calificaciones
- Manualdehigieneindustrialagentesqumicos PDFDocumento71 páginasManualdehigieneindustrialagentesqumicos PDFSusana GizziAún no hay calificaciones
- Introduccion A La Psicologia ClinicaDocumento42 páginasIntroduccion A La Psicologia ClinicaJ C Torres FormalabAún no hay calificaciones
- Guía de Practica de FarmacologíaDocumento64 páginasGuía de Practica de FarmacologíaXavier Perez OdicioAún no hay calificaciones
- Principios - Farmacologia - Grupo152003-26Documento26 páginasPrincipios - Farmacologia - Grupo152003-26Rocio hoyosAún no hay calificaciones
- Farmacologia Libro TEXTODocumento172 páginasFarmacologia Libro TEXTOIdalis RomeroAún no hay calificaciones
- Tecnología Farmacéutica IIDocumento166 páginasTecnología Farmacéutica IIAngely Oviedo RomeroAún no hay calificaciones
- Hemoglobina 1 - Porfirinas. Hierro HZDocumento12 páginasHemoglobina 1 - Porfirinas. Hierro HZyamileAún no hay calificaciones
- Farmacología Gral. Práctica 3 C.PDocumento7 páginasFarmacología Gral. Práctica 3 C.PKelly Espinopsa GamaAún no hay calificaciones
- Diseño de Un Alcoholimetro Digital Acoplado A Un LRD Que Permite Analizar Los Parametros de Frecuencia Cardiaca Utilizando Un Arduino MegaDocumento101 páginasDiseño de Un Alcoholimetro Digital Acoplado A Un LRD Que Permite Analizar Los Parametros de Frecuencia Cardiaca Utilizando Un Arduino MegaDAYANA CAROLINA PARDO DELGADOAún no hay calificaciones
- Trabajo y FarmacologíaDocumento60 páginasTrabajo y FarmacologíaMaribel Chaves MartínezAún no hay calificaciones
- Clase de GastrointestinalDocumento42 páginasClase de GastrointestinalPacora LpacorabAún no hay calificaciones
- Proceso AdmeDocumento2 páginasProceso AdmeDaniela BarbaAún no hay calificaciones
- Farmacocinética y FarmacodinamiaDocumento10 páginasFarmacocinética y FarmacodinamiaLuis PeraltaAún no hay calificaciones
- m15102b03 Acido Folico-5mgDocumento3 páginasm15102b03 Acido Folico-5mgLUISA BERTHA ADUVIRI CORDOVAAún no hay calificaciones
- Informe Practica 1Documento13 páginasInforme Practica 1carla100% (1)
- Farmacocinetica 1Documento24 páginasFarmacocinetica 1Ana.Aún no hay calificaciones
- Adme de MedicamentosDocumento37 páginasAdme de MedicamentosGEORGES JAIR VASQUEZ CHUQUEAún no hay calificaciones
- Clase Magistral FarmacoterapiaDocumento107 páginasClase Magistral FarmacoterapiaMariluz Chura Chambi0% (1)
- FARMACOLOGÍADocumento62 páginasFARMACOLOGÍAEduardo PinedaAún no hay calificaciones
- Farmacología Geriatrica - Uso de MedicamentosDocumento43 páginasFarmacología Geriatrica - Uso de MedicamentosNemo Pumashonco ChávezAún no hay calificaciones
- Conceptos Básicos de FarmacologíaDocumento28 páginasConceptos Básicos de FarmacologíaCATALINA F GUAJARDO MANSILLA100% (1)
- Clase 1 Introduccion y Farmacocinetica PDFDocumento89 páginasClase 1 Introduccion y Farmacocinetica PDFKaren Andrea Soto MillanAún no hay calificaciones
- MKT, Fasting, FedDocumento5 páginasMKT, Fasting, Fedlucia martinezAún no hay calificaciones
- Uso de AntibioticosDocumento15 páginasUso de AntibioticosBrendaCelina MejíaAguayoAún no hay calificaciones
- Taller FARMACODocumento3 páginasTaller FARMACOValentina BuitragoAún no hay calificaciones
- Capitulo 2 FarmacocineticaDocumento62 páginasCapitulo 2 FarmacocineticaPandapandaAún no hay calificaciones
- Farmacocinética Cap 2 y Farmacodinámica Cap3Documento9 páginasFarmacocinética Cap 2 y Farmacodinámica Cap33628986180101Aún no hay calificaciones
- TEMA 3 - Bases Farmacocineticas. Absorcion y DistribucionDocumento40 páginasTEMA 3 - Bases Farmacocineticas. Absorcion y DistribucionLuis VelaAún no hay calificaciones