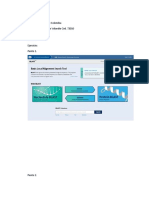
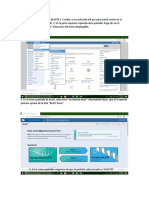
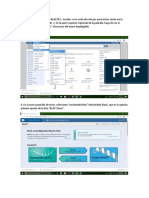
También podría gustarte
- BioinformáticaDocumento4 páginasBioinformáticaNick Kevin Silva G100% (1)
- Ejercicio Bioinformatica 3Documento9 páginasEjercicio Bioinformatica 3Andres VelandiaAún no hay calificaciones
- Taller Alineamiento de Secuencias y Diseño de PrimersDocumento11 páginasTaller Alineamiento de Secuencias y Diseño de PrimersDayanna JaimesAún no hay calificaciones
- Práctico1 2022 EstudiantesDocumento5 páginasPráctico1 2022 EstudiantesAida SanchezAún no hay calificaciones
- Alineamientos PareadosDocumento6 páginasAlineamientos Pareadosfelipe romeroAún no hay calificaciones
- Ejercicios BLASTDocumento4 páginasEjercicios BLAST317088492Aún no hay calificaciones
- Klboncar - Guia Laboratorio 6 BioinformaticaDocumento17 páginasKlboncar - Guia Laboratorio 6 BioinformaticaBartsina FulgenxiaAún no hay calificaciones
- Practica 3 Barcoding Munoz SiguenzaDocumento20 páginasPractica 3 Barcoding Munoz SiguenzaSasha Valeria Siguenza Robles33% (3)
- Formato GenBankDocumento9 páginasFormato GenBankPaola PalaciosAún no hay calificaciones
- Bases de Datos Moleculares 1Documento6 páginasBases de Datos Moleculares 1LEON PARODI LENARDUZZIAún no hay calificaciones
- Práctica N°10-Introducción A La Bioinformática Como Recurso BiotecnológicoDocumento11 páginasPráctica N°10-Introducción A La Bioinformática Como Recurso BiotecnológicoPío OchoaAún no hay calificaciones
- Taller2 Alineam de Secuencias X BLAST y UGENEDocumento11 páginasTaller2 Alineam de Secuencias X BLAST y UGENEccanelounionAún no hay calificaciones
- AndreDocumento10 páginasAndrerodriguezlopezandreAún no hay calificaciones
- Wuolah Free App 1706189836558 Gulag FreeDocumento8 páginasWuolah Free App 1706189836558 Gulag Freejaja jajaAún no hay calificaciones
- Trabajo de BIOINFORMATICADocumento19 páginasTrabajo de BIOINFORMATICANathaly RomeroAún no hay calificaciones
- Vázquez Alejandra P9Documento6 páginasVázquez Alejandra P9aaleev1924234Aún no hay calificaciones
- BioinformáticaDocumento7 páginasBioinformáticaGonzalo Alexis Guíñez YáñezAún no hay calificaciones
- Taller BioinformaticaDocumento8 páginasTaller BioinformaticaPaula RinconAún no hay calificaciones
- MU159 20221 BioEstructural PEC4Documento11 páginasMU159 20221 BioEstructural PEC4gabiAún no hay calificaciones
- Rep FinalDocumento74 páginasRep FinalUlisses Armando TorresAún no hay calificaciones
- Informe N°02 - Informe de Uso Del Software Mega DnaDocumento14 páginasInforme N°02 - Informe de Uso Del Software Mega DnaWendy Hinojosa Ramirez100% (1)
- Practica #14 y 15Documento7 páginasPractica #14 y 15Yanira0% (1)
- Practica 1 GenesDocumento9 páginasPractica 1 GenesMiguelAún no hay calificaciones
- Practica de Laboratorio Bioinformatica 1Documento4 páginasPractica de Laboratorio Bioinformatica 1alesssss100% (1)
- 6 - Manejo Bases de DatosDocumento11 páginas6 - Manejo Bases de DatosNatalia HernandezAún no hay calificaciones
- Examen de BioinformaticaDocumento14 páginasExamen de BioinformaticaJordi Ysmael Camara CabelloAún no hay calificaciones
- Laboratorio 10-Introducción A La BioinformáticaDocumento20 páginasLaboratorio 10-Introducción A La BioinformáticaCarlos D. DueñasAún no hay calificaciones
- Apuntes 2Documento3 páginasApuntes 2JeffersonPalaciosAún no hay calificaciones
- Serial ClonerDocumento7 páginasSerial Clonerkarla figueroa espinosaAún no hay calificaciones
- Unidad1 - AnalisisdeSecuencias - B (3-4)Documento50 páginasUnidad1 - AnalisisdeSecuencias - B (3-4)Micaela AvacaAún no hay calificaciones
- PIA1 Biotecnologia Informática 171Documento26 páginasPIA1 Biotecnologia Informática 171Cristoper Ariel López VenegasAún no hay calificaciones
- Prueba2 EYAG 2022sem2 v1Documento3 páginasPrueba2 EYAG 2022sem2 v1Martín Vergara VargasAún no hay calificaciones
- BioinformaticaDocumento16 páginasBioinformaticaAguedo Huamani HuayhuaAún no hay calificaciones
- Informe de Diseño Primer CompletoDocumento48 páginasInforme de Diseño Primer CompletoMaleh BuitrónAún no hay calificaciones
- Biotecnologia - Tipos y PreguntasDocumento26 páginasBiotecnologia - Tipos y PreguntasLeo BravoAún no hay calificaciones
- Practica 1Documento9 páginasPractica 1Dayana Velarde VigilAún no hay calificaciones
- Seminario 3 PDFDocumento9 páginasSeminario 3 PDFmariaAún no hay calificaciones
- Secuenciacion Del Gen 16sDocumento25 páginasSecuenciacion Del Gen 16sMaria Fernanda AvAún no hay calificaciones
- Elaboración de Dendrogramas A Partir de Artículos Científicos Utilizando El Programa Bioinformático Mega DnaDocumento19 páginasElaboración de Dendrogramas A Partir de Artículos Científicos Utilizando El Programa Bioinformático Mega DnaJosue Calcina Fuentes100% (1)
- Trabajo Algoritmos GeneticosDocumento15 páginasTrabajo Algoritmos GeneticosjulianAún no hay calificaciones
- Reporte BLASTDocumento13 páginasReporte BLASTGustavo López ToledoAún no hay calificaciones
- Metodos Cuantificacion Expresion GenicaDocumento63 páginasMetodos Cuantificacion Expresion GenicaAlfonso Olaya AbrilAún no hay calificaciones
- Guía 4 - Genome Browser2018-2Documento9 páginasGuía 4 - Genome Browser2018-2Fernanda SantanaAún no hay calificaciones
- Tema 7 - Trabajo Con Secuencias APUNTESDocumento19 páginasTema 7 - Trabajo Con Secuencias APUNTESMarcos RodriguezAún no hay calificaciones
- Guía para El Uso de BLASTN 1Documento5 páginasGuía para El Uso de BLASTN 1Luis Castillo CORTESAún no hay calificaciones
- Blast NcbiDocumento6 páginasBlast NcbimarioaAún no hay calificaciones
- Bioinformatica Bioq 2019 CSDocumento20 páginasBioinformatica Bioq 2019 CSJonathan B Gutierrez LeyvaAún no hay calificaciones
- Practica Calificada BioinformaticaDocumento2 páginasPractica Calificada BioinformaticaLuis Eduardo Strater Mc LellanAún no hay calificaciones
- Cuestionario BioinformáticaDocumento3 páginasCuestionario BioinformáticaDominique HerreraAún no hay calificaciones
- Guía para El Uso de BLASTN 1Documento5 páginasGuía para El Uso de BLASTN 1Luis Castillo CORTESAún no hay calificaciones
- Proyecto AI KERASDocumento34 páginasProyecto AI KERASAndrea JuliettAún no hay calificaciones
- Informe Diseño de Epitopes (Inmunologia)Documento6 páginasInforme Diseño de Epitopes (Inmunologia)kevin.lopez.bAún no hay calificaciones
- Tarea 1 Ingeniería MetabólicaDocumento5 páginasTarea 1 Ingeniería MetabólicaNora Torres CastilloAún no hay calificaciones
- Primers Blast (Ncbi)Documento8 páginasPrimers Blast (Ncbi)JuanAún no hay calificaciones
- Taller Eukaryotic Room 3 Ed-Feb-27-2023Documento7 páginasTaller Eukaryotic Room 3 Ed-Feb-27-2023dsescobarmAún no hay calificaciones
- Taller 3Documento9 páginasTaller 3Carlos Israel Esparza AndradeAún no hay calificaciones
- BC Taller 5 BlastDocumento15 páginasBC Taller 5 BlastgeraldineAún no hay calificaciones
- Sesión de Laboratorio No 2.Documento24 páginasSesión de Laboratorio No 2.karla figueroa espinosaAún no hay calificaciones
- Desarrrollo de Una Base de Análisis de Datos. BioinformáticaDocumento51 páginasDesarrrollo de Una Base de Análisis de Datos. BioinformáticaLuis Alberto Marín AguilarAún no hay calificaciones
- Fundamentos de Programación y Bases de Datos: 2ª EdiciónDe EverandFundamentos de Programación y Bases de Datos: 2ª EdiciónAún no hay calificaciones
- Actividad Spsicologia MaquetasDocumento10 páginasActividad Spsicologia MaquetasIngrid Yurley CAICEDO ALVAREZAún no hay calificaciones
- PC-0539-GRL-03 Aislación y Bloqueo Rev4Documento36 páginasPC-0539-GRL-03 Aislación y Bloqueo Rev4rodrigo antonio valencia reyesAún no hay calificaciones
- ESTRUCTURA y Analisis de Inteligencia EmocionalDocumento5 páginasESTRUCTURA y Analisis de Inteligencia EmocionalCarlos Alberto Rojas MolinaAún no hay calificaciones
- Cortes de Agua Del 19 Al 22 Octubre en BogotáDocumento3 páginasCortes de Agua Del 19 Al 22 Octubre en BogotáPRI RADIOTVAún no hay calificaciones
- Teoría Sintética de La Evolución y Fuerzas EvolutivasDocumento21 páginasTeoría Sintética de La Evolución y Fuerzas EvolutivasAndrea :3Aún no hay calificaciones
- Panel Solar AeronavesDocumento3 páginasPanel Solar AeronavesFelixAún no hay calificaciones
- Guía de Brigada de Gestión de Riesgo Yadministracion de Desastre (Autoguardado)Documento22 páginasGuía de Brigada de Gestión de Riesgo Yadministracion de Desastre (Autoguardado)ellys vidalAún no hay calificaciones
- 20135316739manual Sustancias QuimicasDocumento93 páginas20135316739manual Sustancias QuimicasGerardo Salazar Lopez100% (1)
- Enucleación BovinaDocumento3 páginasEnucleación BovinaERIKA ARCE GAVIRIAAún no hay calificaciones
- Oxansil 17 PDFDocumento1 páginaOxansil 17 PDFAspro Brazil Se IglesiasAún no hay calificaciones
- Marvin HarrisDocumento14 páginasMarvin HarrisCristhian Vilca OtazuAún no hay calificaciones
- Preguntas de Evaluación 8Documento2 páginasPreguntas de Evaluación 8Mara PierreAún no hay calificaciones
- Exp. 4378-2018-92Documento12 páginasExp. 4378-2018-92haiden de la palmaAún no hay calificaciones
- Examen TS LDocumento36 páginasExamen TS LmaryteAún no hay calificaciones
- FarmacologiaDocumento32 páginasFarmacologiaOscar VejarAún no hay calificaciones
- Oportunidades de Mejora de SG-SSTDocumento11 páginasOportunidades de Mejora de SG-SSTLinda Tatiana RODRIGUEZ SANCHEZAún no hay calificaciones
- Practica 3 BioquimicaDocumento3 páginasPractica 3 BioquimicaIvis RodriguezAún no hay calificaciones
- Tarea 3 - Los Enfoques Disciplinares en Psicología.Documento39 páginasTarea 3 - Los Enfoques Disciplinares en Psicología.kelly palmaAún no hay calificaciones
- Boletin Semana N°06 - Ciclo Especial de Verano Virtual 2023Documento209 páginasBoletin Semana N°06 - Ciclo Especial de Verano Virtual 2023ggml2647Aún no hay calificaciones
- Enzimas Microbianas para La Industria Textil Lanera N. OliveraDocumento15 páginasEnzimas Microbianas para La Industria Textil Lanera N. OliveraLisseth HuayanayAún no hay calificaciones
- PRÁCTICA 7 PRUEBA PATERNIDAD BMC - pdf.2Documento11 páginasPRÁCTICA 7 PRUEBA PATERNIDAD BMC - pdf.2isaac padillaAún no hay calificaciones
- Geo5 Gavion Reporte SalidaDocumento11 páginasGeo5 Gavion Reporte SalidaManuel Gabriel ConcepciónAún no hay calificaciones
- Manifestaciones Clínicas y Diagnóstico de Pancreatitis AgudaDocumento23 páginasManifestaciones Clínicas y Diagnóstico de Pancreatitis AgudaeddcitoAún no hay calificaciones
- Informe de Transito Estacion El AeropuertoDocumento5 páginasInforme de Transito Estacion El AeropuertoMEGATETOMAún no hay calificaciones
- Charles Spurgeon. Comentario BiblicoDocumento3259 páginasCharles Spurgeon. Comentario BiblicoAlicia Ortiz94% (17)
- Escrito de Acusacion Grupo 12Documento22 páginasEscrito de Acusacion Grupo 12MARIANA ALEJANDRA RAMIREZ CRUZAún no hay calificaciones
- Iluminacion EficazDocumento4 páginasIluminacion EficazRocio PeñaAún no hay calificaciones
- Ficha de Mantenimiento TomacorrientesDocumento1 páginaFicha de Mantenimiento TomacorrientesCacallica Caceres agustoAún no hay calificaciones
- PGG ZId 0 PM XVP Q23 B 85Documento21 páginasPGG ZId 0 PM XVP Q23 B 85Alex CalleAún no hay calificaciones
- 1ra Parte TRAB - ESTADDocumento21 páginas1ra Parte TRAB - ESTADPrieto RicardoAún no hay calificaciones