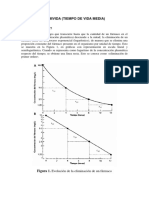
También podría gustarte
- Instrumentación y laboratorio. Manual de procedimientos básicosDe EverandInstrumentación y laboratorio. Manual de procedimientos básicosCalificación: 3 de 5 estrellas3/5 (1)
- Farmacometría:Curvas dosis-respuesta de tipo gradual. Volumen 1De EverandFarmacometría:Curvas dosis-respuesta de tipo gradual. Volumen 1Aún no hay calificaciones
- TIVADocumento70 páginasTIVAAlexey Zamora Betancourt100% (2)
- Toma de muestras y análisis in-situ. QUIE0108: Operaciones básicas en planta químicaDe EverandToma de muestras y análisis in-situ. QUIE0108: Operaciones básicas en planta químicaAún no hay calificaciones
- Tiva Parte 1Documento56 páginasTiva Parte 1pablocotan100% (2)
- Tiva TciDocumento40 páginasTiva TciAnestesiologia Eva Peron100% (9)
- Roctem: Refracción Ocular con Tiempo de Exposición en la MiopizaciónDe EverandRoctem: Refracción Ocular con Tiempo de Exposición en la MiopizaciónAún no hay calificaciones
- Laboratorio de Farmacología, Escuela Superior de Medicina Del IpnDocumento10 páginasLaboratorio de Farmacología, Escuela Superior de Medicina Del IpnBlanca OdetteAún no hay calificaciones
- Ejercicios de Farmacocinética Segunda y Tercera Unidad PDFDocumento6 páginasEjercicios de Farmacocinética Segunda y Tercera Unidad PDFgerson1contreras-1100% (1)
- Recapitulativo Sistemas InyecciónDocumento200 páginasRecapitulativo Sistemas Inyeccióntochas100% (16)
- Clase FarmacocinéticaDocumento29 páginasClase FarmacocinéticaNAGHELI IBETH LLANOS SANCHEZAún no hay calificaciones
- Práctica Vía Oral CorregidaDocumento11 páginasPráctica Vía Oral CorregidaLuis MartinezAún no hay calificaciones
- Práctica No. 8 Estudio Farmacocinético de Safranina Mediante La Utilización de Un Modelo Físico Representativo de Administración IntravenosaDocumento5 páginasPráctica No. 8 Estudio Farmacocinético de Safranina Mediante La Utilización de Un Modelo Físico Representativo de Administración IntravenosaKevin Alexis Madrigal HernandezAún no hay calificaciones
- Práctica 2 - IVDocumento7 páginasPráctica 2 - IVJavier GarcíaAún no hay calificaciones
- Monocompartimental 1Documento11 páginasMonocompartimental 1Luz Deisy Berrios GonzalesAún no hay calificaciones
- Tema 4 Modelo Monocompartimental-Adm Intrav Dosis ÚnicaDocumento11 páginasTema 4 Modelo Monocompartimental-Adm Intrav Dosis ÚnicaRosa María Soto PérezAún no hay calificaciones
- GUIA PRACTICA 4 Modelo MonocompartimentalDocumento4 páginasGUIA PRACTICA 4 Modelo MonocompartimentalAnayanci OrtizAún no hay calificaciones
- Problema Seminario Nº1 Texto (Mono-IV)Documento1 páginaProblema Seminario Nº1 Texto (Mono-IV)Sara SanabriaAún no hay calificaciones
- Practica 5. Simulaci - N Farmacocinética in Vitro de Un Modelo MonocompartimentalDocumento8 páginasPractica 5. Simulaci - N Farmacocinética in Vitro de Un Modelo MonocompartimentalNicolás Carmona100% (1)
- Practica 3 BiofarmaciaDocumento7 páginasPractica 3 BiofarmaciaMiranda VicmontAún no hay calificaciones
- Practica 4Documento35 páginasPractica 4Andrea del Pilar Obeso TantapomaAún no hay calificaciones
- P5 OralDocumento10 páginasP5 OralMoisesAún no hay calificaciones
- Práctica 3 - VODocumento8 páginasPráctica 3 - VOJavier GarcíaAún no hay calificaciones
- P5 OralDocumento10 páginasP5 OralSamuel DoomWyldeAún no hay calificaciones
- MAUC-IV: Simulación de la administración intravenosa de fármacosDocumento7 páginasMAUC-IV: Simulación de la administración intravenosa de fármacosAlbertoAún no hay calificaciones
- Cuestionario Apunte Fundamentos BiofarmaDocumento14 páginasCuestionario Apunte Fundamentos BiofarmaAlfredo JiménezAún no hay calificaciones
- Cuestiones Tipo Test Sobre Fundamentos de Biofarmacia y FarmacocinéticaDocumento13 páginasCuestiones Tipo Test Sobre Fundamentos de Biofarmacia y FarmacocinéticaJosé Vélez100% (1)
- EXAMEN EVALFARMA Ordinario B APLICAR 26-05-20 OKDocumento7 páginasEXAMEN EVALFARMA Ordinario B APLICAR 26-05-20 OKMarlon GuerreroAún no hay calificaciones
- Práctica - Mauc EvDocumento4 páginasPráctica - Mauc EvAlejandro Parada FloresAún no hay calificaciones
- Practica No 9 Problemasmod Comp y No Comp.Documento9 páginasPractica No 9 Problemasmod Comp y No Comp.Valeery CruzAún no hay calificaciones
- Modelo abierto de administración en dosis repetidasDocumento6 páginasModelo abierto de administración en dosis repetidasMario MaldonadoAún no hay calificaciones
- Aplicaciones MatemáticasDocumento9 páginasAplicaciones MatemáticasEduardo Leura100% (1)
- Práctica 5 VODocumento13 páginasPráctica 5 VOJosseline SuarezAún no hay calificaciones
- Reporte P2. Farmacocinética IDocumento8 páginasReporte P2. Farmacocinética IHugo ValedoAún no hay calificaciones
- INFORME 2 - CINETICA FinalDocumento15 páginasINFORME 2 - CINETICA FinalTHALIA YESSENIA APONTE DE LA CRUZAún no hay calificaciones
- Practica Farmacocinetica IDocumento7 páginasPractica Farmacocinetica IepikexdeeAún no hay calificaciones
- FarmacocineticaDocumento9 páginasFarmacocineticaFabian PinitoAún no hay calificaciones
- FarmacocinéticaDocumento23 páginasFarmacocinéticaDominic Guerra RíosAún no hay calificaciones
- Analisis CompartimentalDocumento51 páginasAnalisis CompartimentalMiguel Atzin Pozos100% (2)
- Farma 1 Practica 8Documento6 páginasFarma 1 Practica 8Roxana HernandezAún no hay calificaciones
- Modelo Abierto de Un Compartimiento Admón IVDocumento61 páginasModelo Abierto de Un Compartimiento Admón IVDIANA ALLISON QUINONES JAUREGUIAún no hay calificaciones
- Caso Clínico Real para El Cálculo de Un Inyección Con Ecuaciones Diferenciales OrdinariasDocumento17 páginasCaso Clínico Real para El Cálculo de Un Inyección Con Ecuaciones Diferenciales OrdinariasJinhichi Molero RodriguezAún no hay calificaciones
- TP4 cinética 2 subido a campus 2024Documento11 páginasTP4 cinética 2 subido a campus 2024rochitrimarco12Aún no hay calificaciones
- Farmacocinética clínica: preguntas y conceptos fundamentalesDocumento5 páginasFarmacocinética clínica: preguntas y conceptos fundamentalesAlumnos4C UjedAún no hay calificaciones
- Presentacion Modelo (ORIGINAL)Documento23 páginasPresentacion Modelo (ORIGINAL)Mimi TorresAún no hay calificaciones
- Administración continua de fármaco: Estado estacionario, tiempo medio de residencia y dosis múltipleDocumento14 páginasAdministración continua de fármaco: Estado estacionario, tiempo medio de residencia y dosis múltipleMary Elizabeth ChambiAún no hay calificaciones
- DiscusionDocumento5 páginasDiscusionAlexandraParedesPuertaAún no hay calificaciones
- Sesion 09. Biofarmacia y Farmacocinetica Guia de Ejercicios N°03 Ii UnidadDocumento6 páginasSesion 09. Biofarmacia y Farmacocinetica Guia de Ejercicios N°03 Ii Unidadgerson1contreras-1Aún no hay calificaciones
- Modelo Abierto MonocompartimentalDocumento13 páginasModelo Abierto MonocompartimentalJonathan Molina AguileraAún no hay calificaciones
- Anestesia Total Intravenosa Con Remifentanyl en PediatriaDocumento16 páginasAnestesia Total Intravenosa Con Remifentanyl en PediatriaAndrea Elizabeth Ruíz HérculesAún no hay calificaciones
- Modelo monocompartimental intravenosoDocumento10 páginasModelo monocompartimental intravenosoRosa SotoAún no hay calificaciones
- Farmacocinética: Absorción, Distribución y Eliminación de FármacosDocumento8 páginasFarmacocinética: Absorción, Distribución y Eliminación de FármacossasaAún no hay calificaciones
- Cálculo de Los Parámetros Farmacocinéticos en El Modelo MonocompartimientalDocumento6 páginasCálculo de Los Parámetros Farmacocinéticos en El Modelo Monocompartimientalmargarikilla3164Aún no hay calificaciones
- Practica No 9 Problemasmod Comp y No Comp.Documento3 páginasPractica No 9 Problemasmod Comp y No Comp.Diego Albes Sandoval LopezAún no hay calificaciones
- Exposición BiofarmaciaDocumento10 páginasExposición BiofarmaciaSthefany Saavedra ArgomedoAún no hay calificaciones
- Problemas BiofaDocumento38 páginasProblemas BiofaCarolina LopezAún no hay calificaciones
- Semivida - ModuloDocumento6 páginasSemivida - ModuloLeonardo Rafael Martinez HuapayaAún no hay calificaciones
- Cuestionario Practica 2Documento3 páginasCuestionario Practica 2Ivón Flores100% (1)
- Modelo farmacocinético monocompartimental digoxinaDocumento6 páginasModelo farmacocinético monocompartimental digoxinaluzAún no hay calificaciones
- FARMACOCINETICA Y GESTACIÓN Mg. Hugo JustilDocumento29 páginasFARMACOCINETICA Y GESTACIÓN Mg. Hugo JustilMISHEL ESPINOZA OLIVARESAún no hay calificaciones
- Manual Provisión Manejo y Consumo Alimentación Escolar0173414001633982582Documento13 páginasManual Provisión Manejo y Consumo Alimentación Escolar0173414001633982582Maria ReateguiAún no hay calificaciones
- Entomologia ForenseDocumento16 páginasEntomologia ForenseHumbeto TrejoAún no hay calificaciones
- C-037 Del 2000Documento25 páginasC-037 Del 2000kamilohbkAún no hay calificaciones
- Accidente de Trabajo Trámites Anteriores Al Inicio de Las Acciones Ante Las Comisiones Médicas Regimen Riesgo Del TrabajoDocumento9 páginasAccidente de Trabajo Trámites Anteriores Al Inicio de Las Acciones Ante Las Comisiones Médicas Regimen Riesgo Del TrabajoMartin ToledoAún no hay calificaciones
- Municipio de Suaza RESERVASDocumento8 páginasMunicipio de Suaza RESERVASNATALIA SOTELO GOMEZAún no hay calificaciones
- Avances Capitulo I, II y III Jesus TabordaDocumento51 páginasAvances Capitulo I, II y III Jesus Tabordajesus tabordaAún no hay calificaciones
- Gq-Ai-Fo-051 - Proporcion de Cancelacion de CirugiaDocumento1 páginaGq-Ai-Fo-051 - Proporcion de Cancelacion de CirugiaWILSON MAURICIO QUIZA CABRERAAún no hay calificaciones
- Presión aire tanqueDocumento4 páginasPresión aire tanqueEdwin Alonso Guevara BecerraAún no hay calificaciones
- 767 677 PBDocumento189 páginas767 677 PBAndrés Gutiérrez MoralesAún no hay calificaciones
- Tratados comerciales internacionales ZacatecasDocumento3 páginasTratados comerciales internacionales ZacatecasJuan Carlos Alvarez SalazarAún no hay calificaciones
- Caso Práctico Comitas ComunicacionesDocumento2 páginasCaso Práctico Comitas ComunicacionesBlitZ KrieGAún no hay calificaciones
- Laguna de La CochaDocumento21 páginasLaguna de La CochaMonicaAún no hay calificaciones
- Cuadernillo 30 Al 3 de FebDocumento5 páginasCuadernillo 30 Al 3 de FebEH AGAún no hay calificaciones
- Fichas BibliográficasDocumento5 páginasFichas BibliográficasVìctor Quiroz SalazarAún no hay calificaciones
- 1anexo Exp 02 1act Ciencia y Tecnología 5°Documento3 páginas1anexo Exp 02 1act Ciencia y Tecnología 5°Ana Edith Linares CabreraAún no hay calificaciones
- CIMENTACIONES I Strain GagesDocumento26 páginasCIMENTACIONES I Strain GagesDiego Dleon MaNiAún no hay calificaciones
- Hasta La RaízDocumento1 páginaHasta La RaízKaren FerreyraAún no hay calificaciones
- Reglas de Kirchhoff en circuito eléctricoDocumento10 páginasReglas de Kirchhoff en circuito eléctricoFranko Pacheco PerezAún no hay calificaciones
- El Hombre UnidimensionalDocumento2 páginasEl Hombre UnidimensionalIvan VanegasAún no hay calificaciones
- 2012 Folleto STOP EsDocumento4 páginas2012 Folleto STOP Esluiseduardo_plcAún no hay calificaciones
- Criollo Haitiano - Wikipedia, La Enciclopedia Libre PDFDocumento67 páginasCriollo Haitiano - Wikipedia, La Enciclopedia Libre PDFFabián Fabián Pérez perezAún no hay calificaciones
- Secado por atomización de extracto de hojas de eucaliptoDocumento4 páginasSecado por atomización de extracto de hojas de eucaliptoClaudia Llerena CalderónAún no hay calificaciones
- Muebles y accesorios para el hogarDocumento12 páginasMuebles y accesorios para el hogarElizabeth Najarro GonzalesAún no hay calificaciones
- Los efectos de la marihuana en la salud mental y físicaDocumento3 páginasLos efectos de la marihuana en la salud mental y físicaAngel Agustin Marinez HernandezAún no hay calificaciones
- Trabajo 03Documento3 páginasTrabajo 03Carmen AragonAún no hay calificaciones
- Capitulo 7 Terapia Racional Emotiva Conductual (TREC)Documento2 páginasCapitulo 7 Terapia Racional Emotiva Conductual (TREC)anyelierika002Aún no hay calificaciones
- Mi Kpaw 01 SP en FR PT 1Documento41 páginasMi Kpaw 01 SP en FR PT 1Ignacio MonederoAún no hay calificaciones
- Cortado de icopor en máquina artesanalDocumento5 páginasCortado de icopor en máquina artesanalJose Luis Beltran Timote0% (1)
- Diseño Tipográfico Clasificación Fran CruzDocumento4 páginasDiseño Tipográfico Clasificación Fran CruzkarmonasAún no hay calificaciones