También podría gustarte
- Porfiria Eritropoyética Congénita-Caso Clínico-Grupo 1-P3Documento11 páginasPorfiria Eritropoyética Congénita-Caso Clínico-Grupo 1-P3Majo Palacios LopezAún no hay calificaciones
- PsicometriaDocumento2 páginasPsicometriaMajo Palacios LopezAún no hay calificaciones
- Materialism oDocumento27 páginasMaterialism oAgustín TarifaAún no hay calificaciones
- Derechos Sexuales y ReproductivosDocumento2 páginasDerechos Sexuales y ReproductivosMajo Palacios LopezAún no hay calificaciones
- Gestion de EmpresasDocumento20 páginasGestion de EmpresasMajo Palacios LopezAún no hay calificaciones
- Soy Madre y Quiero Educación Sexual y de Género para Mis HijosDocumento4 páginasSoy Madre y Quiero Educación Sexual y de Género para Mis HijosDayana FernándezAún no hay calificaciones
- ITU and Pregnancy AMS-2016.españolDocumento11 páginasITU and Pregnancy AMS-2016.españolMajo Palacios LopezAún no hay calificaciones
- Vaginitis - ACOG 2019.en - Es PDFDocumento12 páginasVaginitis - ACOG 2019.en - Es PDFMajo Palacios LopezAún no hay calificaciones
- Vaginitis - ACOG 2019.en - Es PDFDocumento12 páginasVaginitis - ACOG 2019.en - Es PDFMajo Palacios LopezAún no hay calificaciones
- 1.2 Estrategias para Prescribir AspirinaDocumento7 páginas1.2 Estrategias para Prescribir AspirinaJosselyn ReinaAún no hay calificaciones
- 2.1 Anticoncepción de EmergenciaDocumento7 páginas2.1 Anticoncepción de EmergenciaMajo Palacios LopezAún no hay calificaciones
- A 11 V 18 N 2Documento9 páginasA 11 V 18 N 2Jose MendozaAún no hay calificaciones
- 4.1 Anemia en EmbarazadasDocumento7 páginas4.1 Anemia en EmbarazadasMajo Palacios LopezAún no hay calificaciones
- LactanciaMaternaUniversidadCentralEcuadorDocumento15 páginasLactanciaMaternaUniversidadCentralEcuadorMajo Palacios LopezAún no hay calificaciones
- Trastorno AdaptativoDocumento13 páginasTrastorno AdaptativoMajo Palacios LopezAún no hay calificaciones
- Hueso Nasal y Flujo TricuspideoDocumento9 páginasHueso Nasal y Flujo TricuspideoMajo Palacios LopezAún no hay calificaciones
- EPP entrega formatoDocumento2 páginasEPP entrega formatoluisAún no hay calificaciones
- Terminología Medica - Sistema NerviosoDocumento26 páginasTerminología Medica - Sistema NerviosoJose Benjamin Almanza Orozco100% (1)
- LIBROCURSOPATOLOGABSICADELACOLUMNA2014Documento169 páginasLIBROCURSOPATOLOGABSICADELACOLUMNA2014Ramón ValeraAún no hay calificaciones
- ESTUDIO DE CASO. Ficha 1 (2) (Autoguardado) ListaDocumento2 páginasESTUDIO DE CASO. Ficha 1 (2) (Autoguardado) ListajessicaAún no hay calificaciones
- Parejas Liberales Ayuda Desplazo en MadridDocumento2 páginasParejas Liberales Ayuda Desplazo en MadridEroticoAún no hay calificaciones
- Corn Ec 2012Documento15 páginasCorn Ec 2012Nayelli Farías.Aún no hay calificaciones
- Encuesta de Ansiedad InfantilDocumento1 páginaEncuesta de Ansiedad InfantilJuli QuirozAún no hay calificaciones
- SFSDFDocumento11 páginasSFSDFVid Castillo RamírezAún no hay calificaciones
- Potenciales EvocadosDocumento7 páginasPotenciales Evocadoshebe1985Aún no hay calificaciones
- GERENCIA SOCIAL 2020-I - Semana 4Documento20 páginasGERENCIA SOCIAL 2020-I - Semana 4Carmen CruzAún no hay calificaciones
- Informe Farmacotecnia Jarabe Base y PomadaDocumento18 páginasInforme Farmacotecnia Jarabe Base y PomadaKety León MoyaAún no hay calificaciones
- 11 Una Interrogación Sobre La Dependencia - C. OlievensteinDocumento6 páginas11 Una Interrogación Sobre La Dependencia - C. OlievensteinFederico DanielAún no hay calificaciones
- Mapa ConceptualDocumento2 páginasMapa ConceptualJose Efrain García López OrjuelaAún no hay calificaciones
- Alerta REVITAPROSTDocumento3 páginasAlerta REVITAPROSTDano AcevesAún no hay calificaciones
- PTS - Arenado de Silo, Cambio de PlanchaDocumento14 páginasPTS - Arenado de Silo, Cambio de PlanchaPREVENCIÓN SERVICIOS DE AIRE SACAún no hay calificaciones
- Actividad 4 ContestadaDocumento4 páginasActividad 4 ContestadaXtian Alejandro Hernandez Orduña0% (9)
- Plan Intervencion Essalud 2022 Gg1.0Documento21 páginasPlan Intervencion Essalud 2022 Gg1.0José A Tasayco SeminarioAún no hay calificaciones
- Estrategias de resolución de problemas mediante diagramas de Venn para tutoría de evaluación especialDocumento5 páginasEstrategias de resolución de problemas mediante diagramas de Venn para tutoría de evaluación especialPeter DahintenAún no hay calificaciones
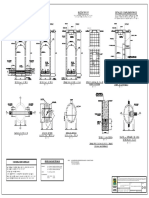
- Detalle de Armadura de Buzones-Bz - TipicosDocumento1 páginaDetalle de Armadura de Buzones-Bz - TipicosJulio Cesar Cordova ReyesAún no hay calificaciones
- Generación de energías limpias MéxicoDocumento7 páginasGeneración de energías limpias MéxicoCanal que uso como nube xd Don't mind itAún no hay calificaciones
- Incapacidad e Historia SanitasDocumento7 páginasIncapacidad e Historia SanitasAshley PinesAún no hay calificaciones
- Pregunta, Hipotesis y ObjetivosDocumento8 páginasPregunta, Hipotesis y ObjetivosRenzo Omar Barreto AbantoAún no hay calificaciones
- 7 - 08 - 2021 10 - 29 - 47 A. M.reporte - AutorizacionDocumento3 páginas7 - 08 - 2021 10 - 29 - 47 A. M.reporte - AutorizacionIVAN DARIO CONDEAún no hay calificaciones
- Ciclo de Volumen y Definición para Mujeres Oxandrolona & PrimobolanDocumento2 páginasCiclo de Volumen y Definición para Mujeres Oxandrolona & PrimobolanDavid Talavera Talavera ReynosoAún no hay calificaciones
- Hemorragia Pulmonar Inducida Por El Ejercicio en El Caballo Una RevisiónDocumento11 páginasHemorragia Pulmonar Inducida Por El Ejercicio en El Caballo Una RevisióncasanchezisAún no hay calificaciones
- Lab. Pecuarios, Productos Procesados CárnicosDocumento11 páginasLab. Pecuarios, Productos Procesados CárnicosANDREA LUCELLY ZULETA LEONAún no hay calificaciones
- Ejemplos de Oraciones Con Conectores CausalesDocumento4 páginasEjemplos de Oraciones Con Conectores Causalesjose miguel Reyes SanchezAún no hay calificaciones
- Teoría Existencialista de Rollo MayDocumento12 páginasTeoría Existencialista de Rollo MayCarlos Zatarain Andrade100% (1)
- ASER-Clase 4Documento30 páginasASER-Clase 4Hugo DonatoAún no hay calificaciones
- Comunicacion en Los Ambitos Escolar y ProfesionalDocumento59 páginasComunicacion en Los Ambitos Escolar y Profesionalfatima cantoAún no hay calificaciones